for the spectra. (I can also share the DICOM file).
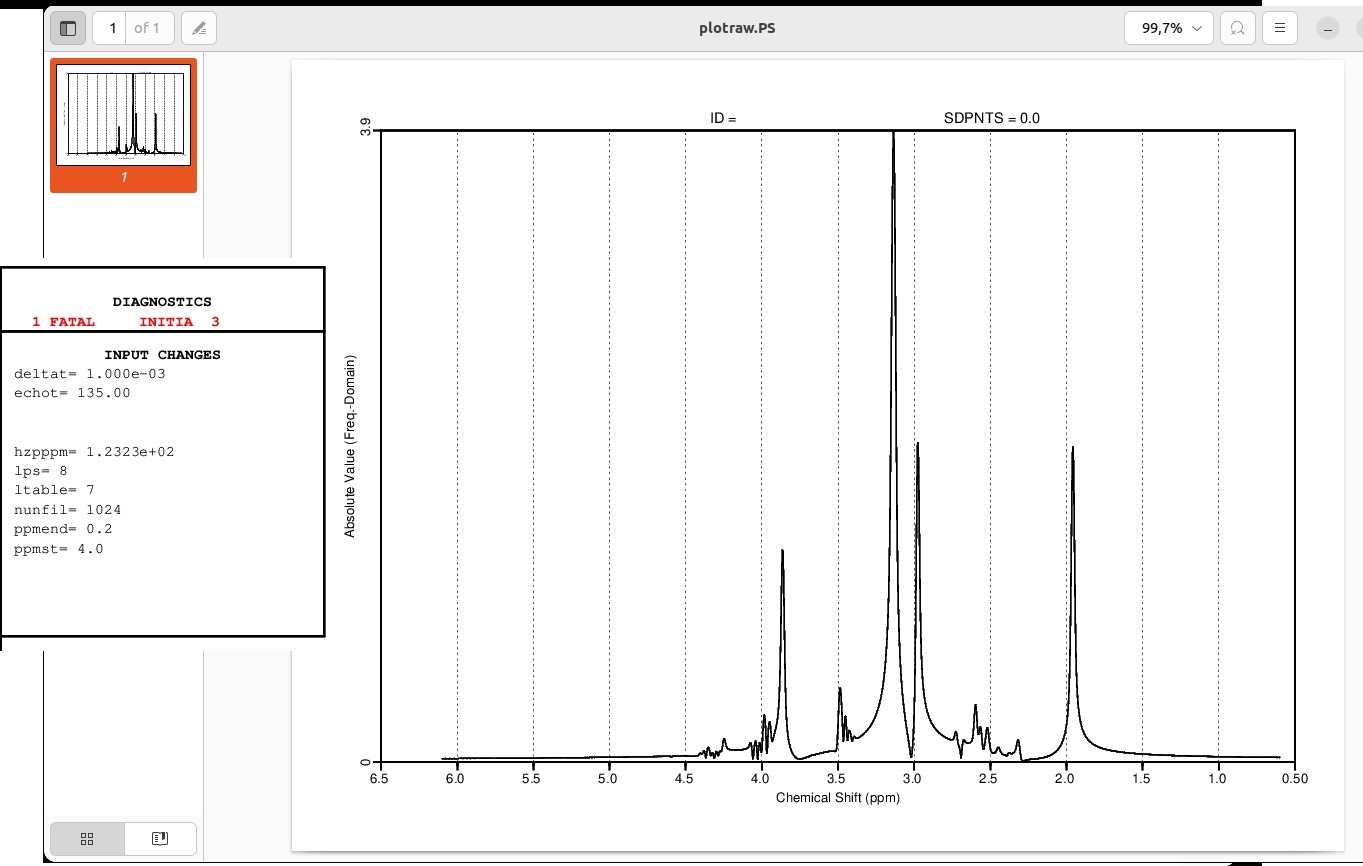
However I am not able to fit it and get error INITIA 3. I’ve tried changing the ppmend and ppmst values but no luck. Maybe someone knows what I may be doing wrong?
To what values did you change PPMST and PPMEND? You can see that the low end of your ppm axis appears to be at around .6 ppm or so. You can always increase the bandwidth of your simulated data, too.
The Cr/Cho section of your simulated data looks a little odd - what do the individual basis functions look like?
The simulation was done using NMR-scope-B with the Cr:Cho:NAA ratio of 1:1:1. These are the initial concentrations I think.
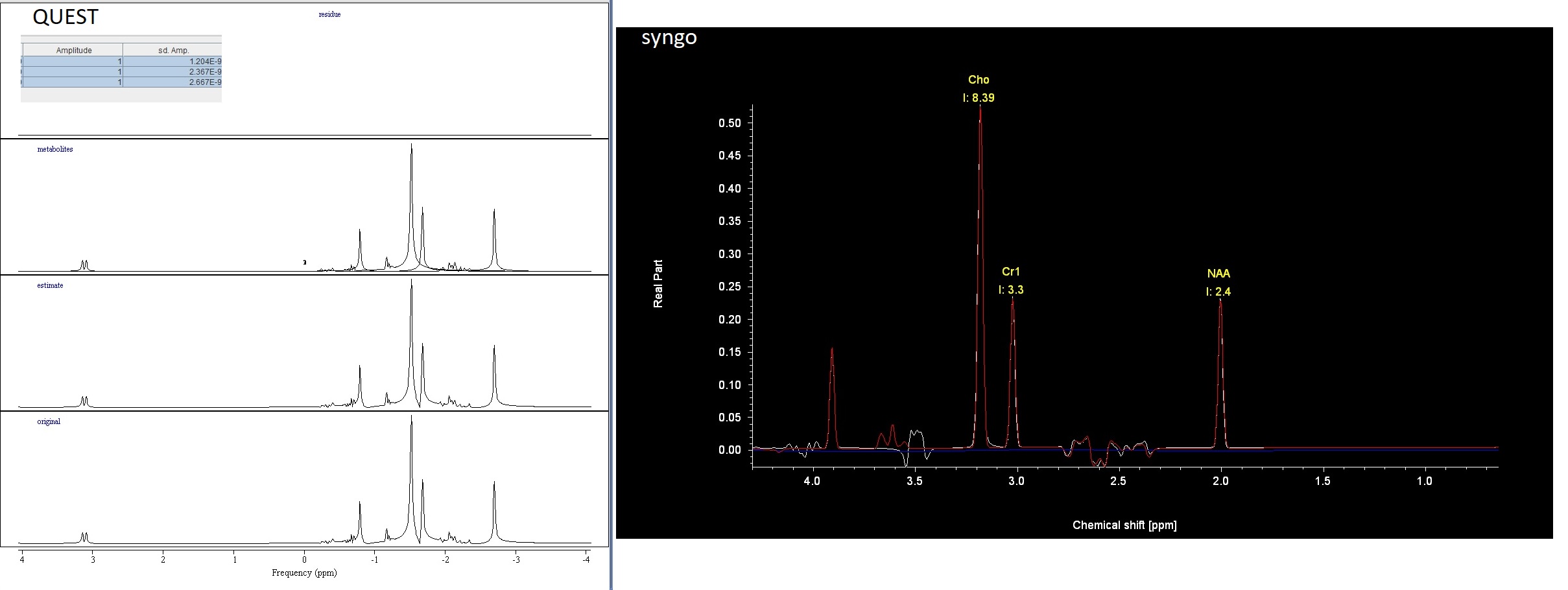
Because I also want to fit simulated data with a program that can only import Siemens DICOM files I acquired a spectro scan with F0 on water, and overwrote the data with my simulated data. example_overwrite_DICOM.m (1.8 KB)
I am then able to fit this in JMRUI-QUEST (perfectly because I’m using the same metabolite models), and in Syngo:
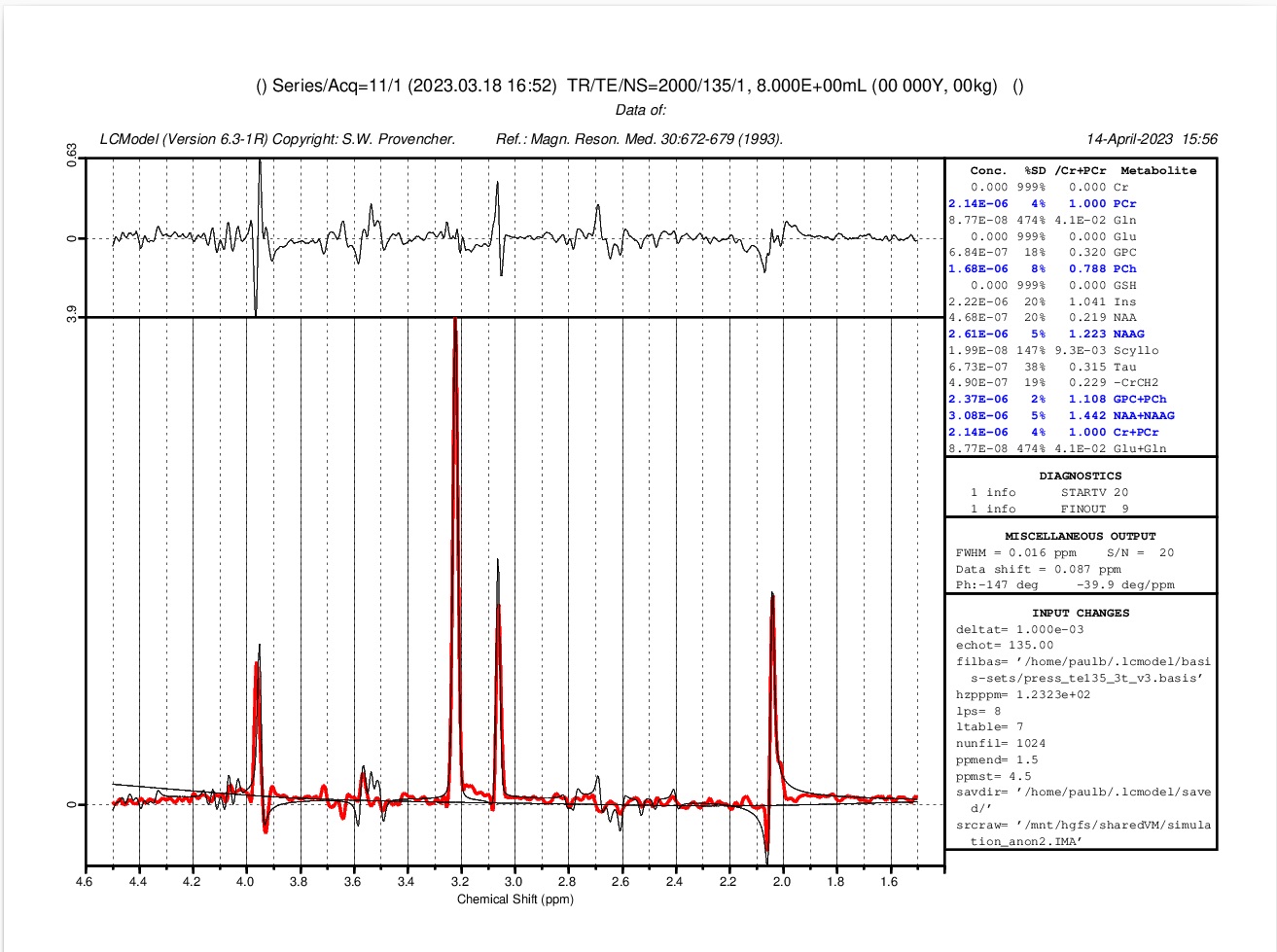
But no luck with LCmodel. If I increase the ppmst to 0.6 I get another error: startv 20 and startv 22. I’ve also tried adding a water peak but no luck. Maybe my spectro is just not realistic enough. For the basis set I’m using press_te135_3t_v3.basis, provided on the LCmodel webpage. I’ll also try your suggestion of a larger BW…
My goal at the moment is just to learn the basics of MR spectroscopy, and I thought simulating a spectra and trying to fit it with different programs would be nice to try. Maybe add noise, baselines, etc later. We also have a more global aim to find an alternative spectroscopy program to replace our current one.
I’ll see if I can make the lc basis set with io_writelcmraw…
After saving the jmrui-NMR-scope-B simulated data as a .m file, I used io_writelcmraw to create the metabolite list: makebase.m (701 Bytes)
and modified your example file to make a .in file: makebasis.txt (644 Bytes)
params I’m unsure about:
mystruct.linewidth
mystruct.t (why in LCmodel basis file this goes negative)
fwhmba=.013 (in .in file)
when using:
$HOME/.lcmodel/bin/makebasis < makebasis.in
I get error:
Fortran runtime error: Cannot match namelist object name cho
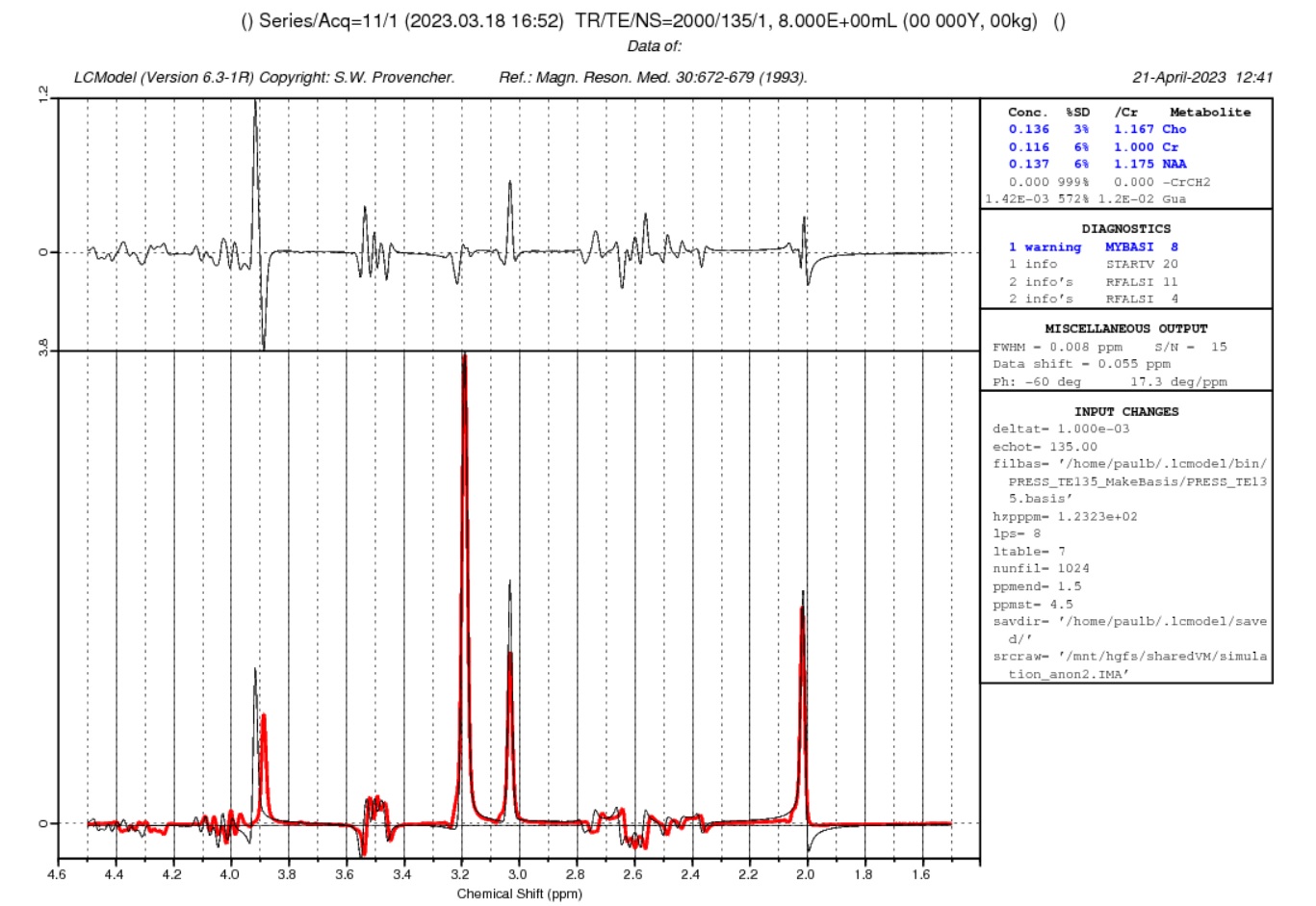
I’m getting close to getting the same basis in LCmodel that I simulated in NMR-scope-B. But it is mirrored in the frequency direction. Is this because of an unwanted complex conjugate?
Thanks. The basis spectra now look good, but the fit is still not perfect. I’ll need to look at every step again.
simulation in NMR-scope-B → *.m file → add all metabolite fids → [overwrite DICOM file with simulated fid]
same *.m file → create LCmodel basis file.
-confirmed dicom file delta_t tag same as simulated delta_t
-exact same B0. x.xx
-only difference I see, is that I simulated with a very large TR, and my dicom file has a TR=2s
-with jrmui-QUEST and Siemens syngo I can fit the spectra-dicom file perfectly. I must be doing something wrong using LCmodel?