@wclarke Thank you for your reply.Below is my processing flow and the issues I encountered. I am very willing to provide you with the original data if necessary.
1.data information
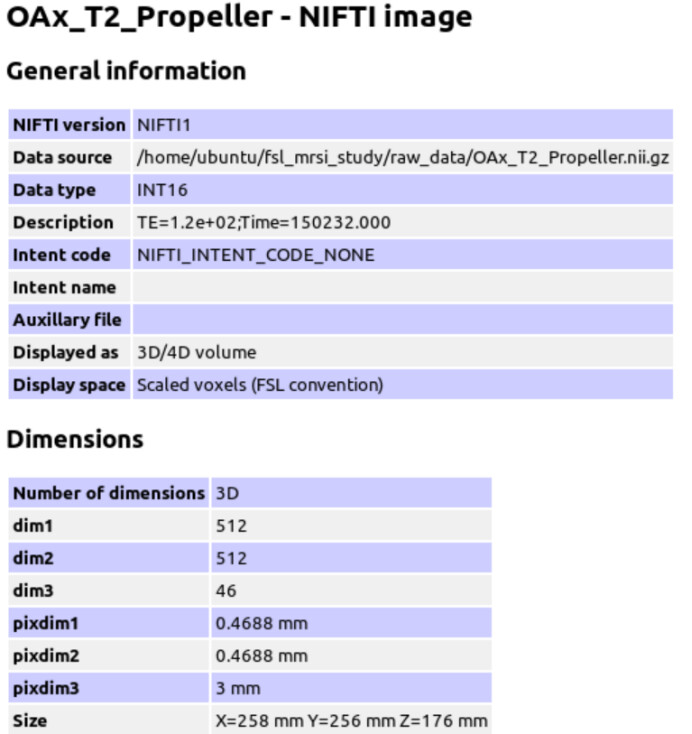
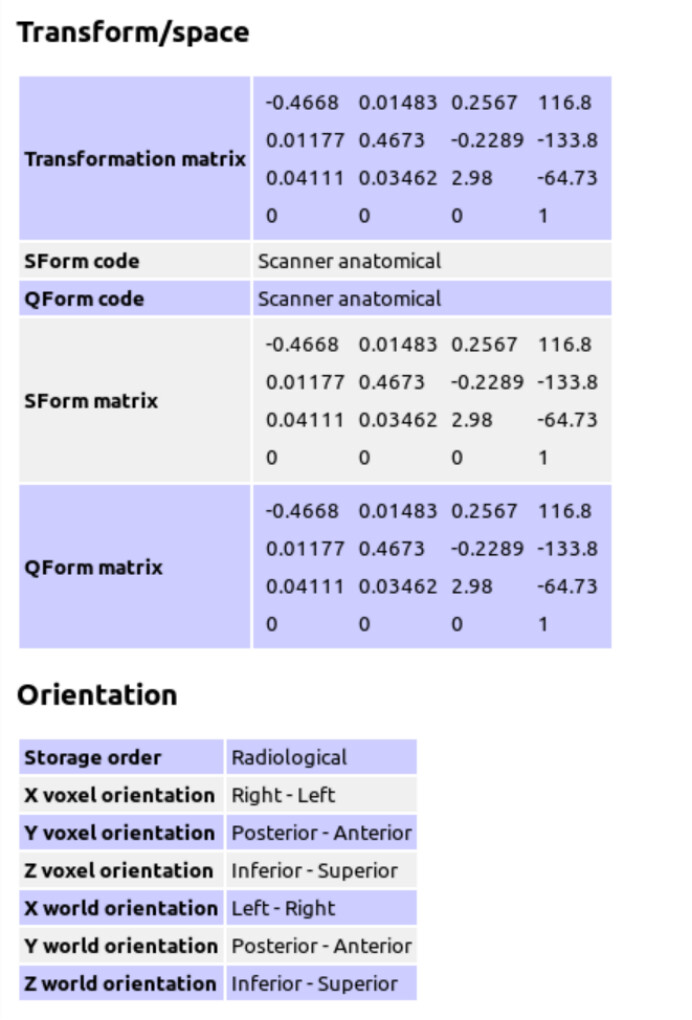
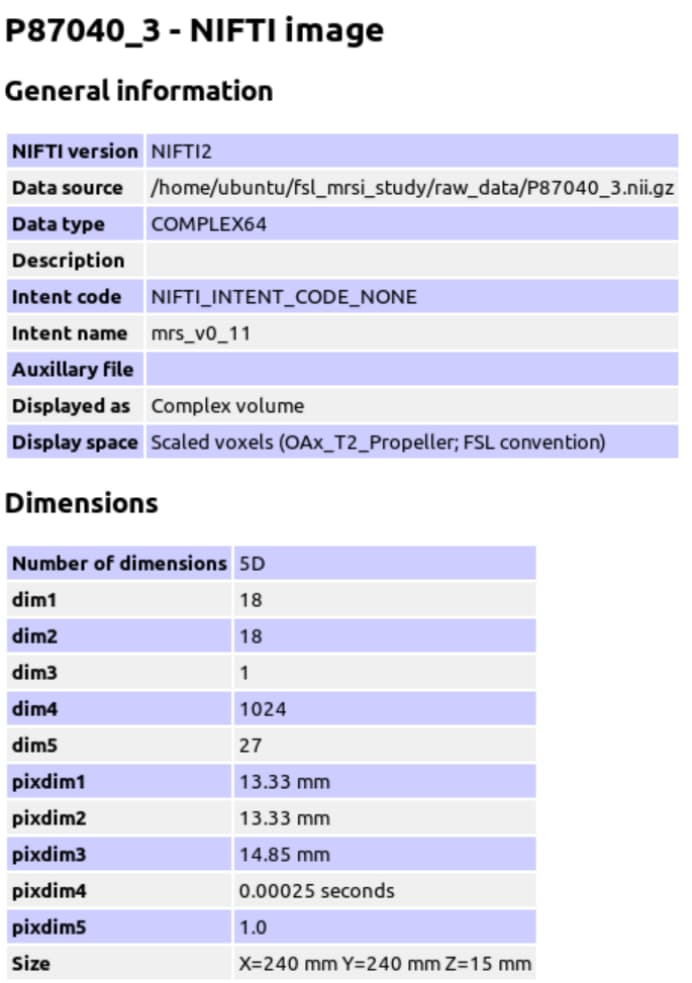
The GE P file is converted to Nifti format using spec2nii to generate the header JSON information.
P87040.txt (971 Bytes)
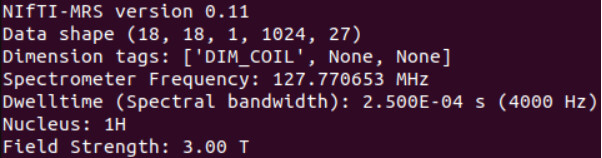
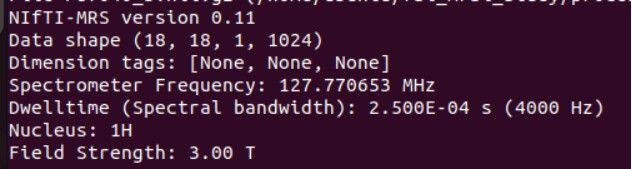
mrs_tools info raw_data/P87040.nii.gz
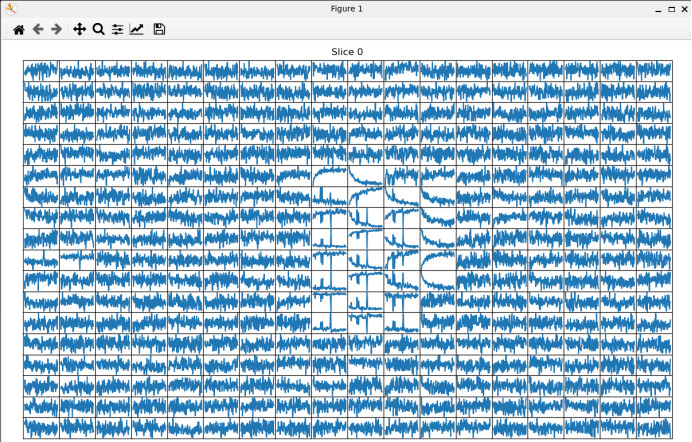
mrs_tools vis raw_data/P87040.nii.gz
2.preprocessing
According to the FSL-MRS documentation(Processing — FSL-MRS 2.4.13 documentation; Quick Start Guide — FSL-MRS 2.4.13 documentation), it was recommended to perform coil-combined and repetitions averaged before fitting, along with specialized preprocessing tools such as inter voxel alignment, phase correction, and lipid removal.
Therefore, I performed coil-combined before fitting only.
Mrs_tools info processed_data/combined/P87040.nii.gz
3.Create Basis Spectra
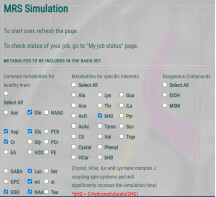
For the metabolite base set, I used MRSCloud on the MRICloud platform to generate it (https://braingps.mricloud.org/mrs-cloud). The metabolite selection and parameter settings are shown in the figure.
parameter settings
Localization:PRESS,sLASER(I choose PRESS)
Vendor:GE
Editing:unedited,MEGA,HERMES,HERCULES(I choose ubedited)
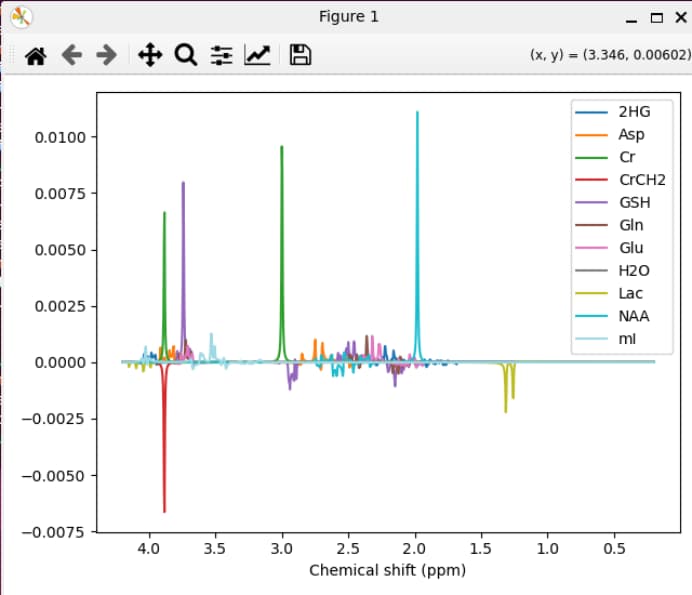
mrs_tools vis GE_PRESS_3T_144ms_fsl
4.Tissue Segmentation
Because the T2 sequence was used as the anatomical reference when performing MRS, I performed tissue segmentation on the T2 sequence.
5.Fitting
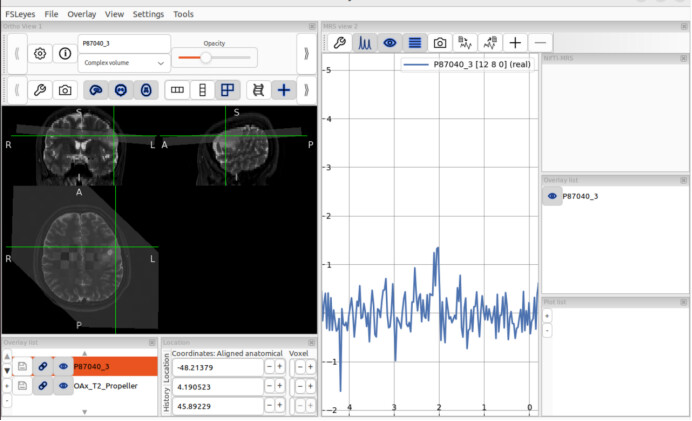
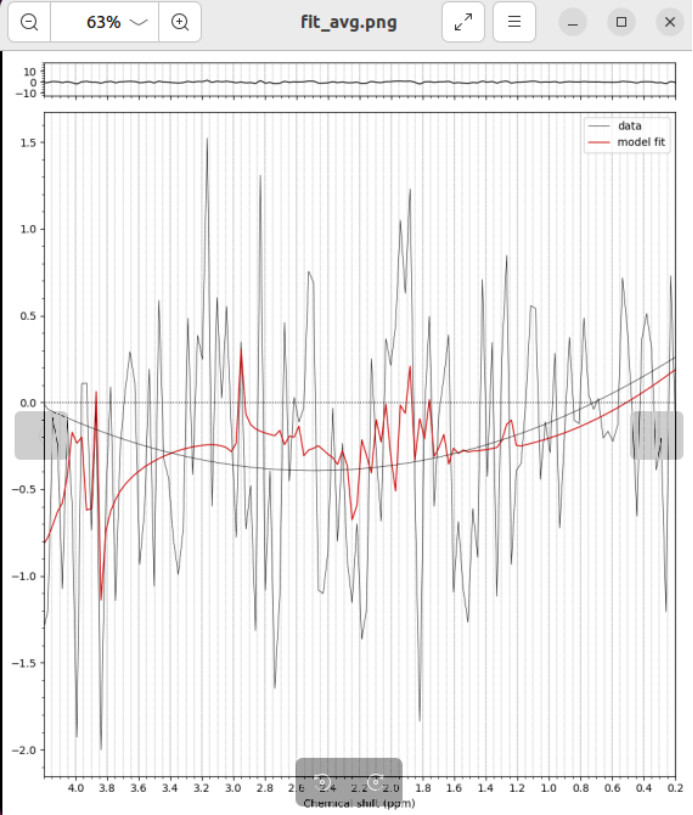
I fitted the coil-combined MRSI data with tissue segmented by T2 sequences.
fsl_mrsi --data processed_data/combined/P87040.nii.gz --basis GE_PRESS_3T_144ms_fsl --output results/P87040 --tissue_frac mrsi_seg_wm.nii.gz mrsi_seg_gm.nii.gz mrsi_seg_csf.nii.gz --internal_ref Cr --report
6.Visualise
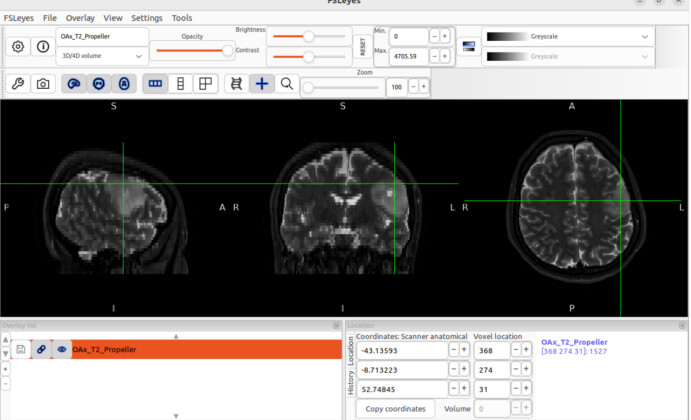
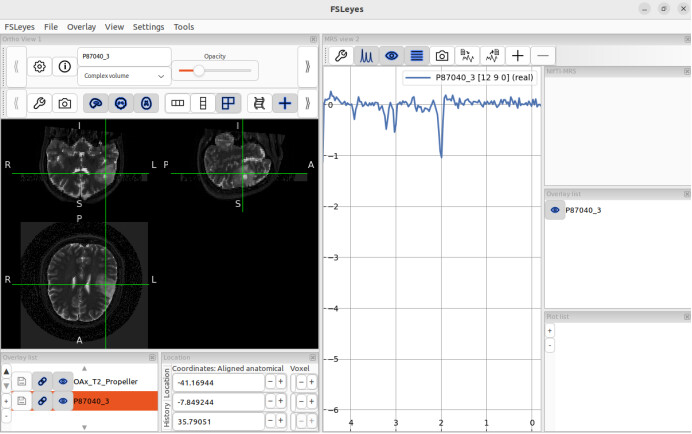
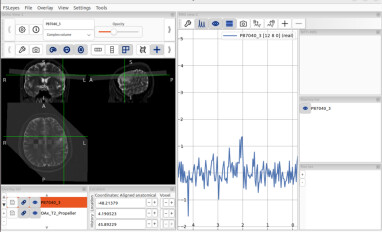
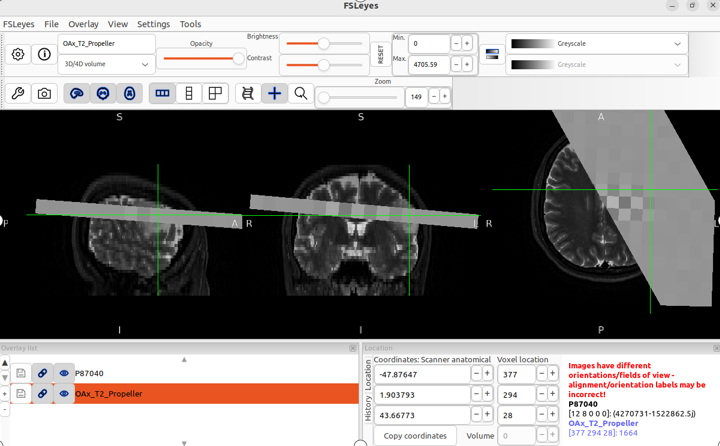
fsleyes -smrs processed_data/combined/P87040.nii.gz raw_data/OAx_T2_Propeller.nii.gz &
When I open fsleyes separately and then load the T2 sequence and MRSI, the positions are displayed correctly, but this is not the case when opening these two files using the command line.
7.My questions.
Is there a problem with my processing for this fitting result?
Is it a problem with the metabolite baseline set parameter settings?
Is it a problem with the MRSI preprocessing?
I’m completely clueless.
I even tried adding several separate preprocessing steps based on MRSI recommendations, but the results were still unsatisfactory.
I also tried T1 sequences for tissue segmentation, but the fitting result was the same.
I would be extremely grateful if someone could offer me some guidance.