A couple of questions here on the fsl-mrs software. I started running some pilot data through fsl-mrs and have run into a small issue when visualizing the data via mrs_vis. I can load the data in fsleyes (super cool )but when running the mrs_vis command I get this error:
Something with the misc.py script… any suggestions on how to fix?
Issue number 2 is in the svs_segment command, I am comparing this pipeline to our own. For the voxel segmentation values, we see quite different results, we use BET and FAST from FSL and get WM=0.19697396 GM=0.58993709 CSF= 0.21308888 for an ~8ML ACC voxel and in fsl-mrs the values are= “CSF”: 0.296241, “GM”: 0.523948, “WM”: 0.179812. Is there anyway to not run the linear registration in svs_segment to compare these a bit more closely?
Lastly, just some visual issues in fsleyes, I installed the latest version and the brain display apears to be tilted back, as if the subject is taking a drink . The file is fine, when I visualize it in MRIcroGL or Matlab, but just curious if other are running into this issue as well. This occurs when I load the unsegmented 3DT1 as well as the segmented and betted files.
Thanks for giving FSL a go! Great to have feedback. It looks like for whatever reason the central frequency hasn’t been picked up in your data. How did the data get generated? Through spec2nii? What do you get if you run mrs_info path/to/your/data.nii.gz? That might also crash, in which case if you use the most recent version of spec2nii you can run spec2nii dump path/to/your/data.nii.gz and it will print what is in the headers.
For the second issue, that might take a bit more figuring out. Which bit of linear registration are you referring to?
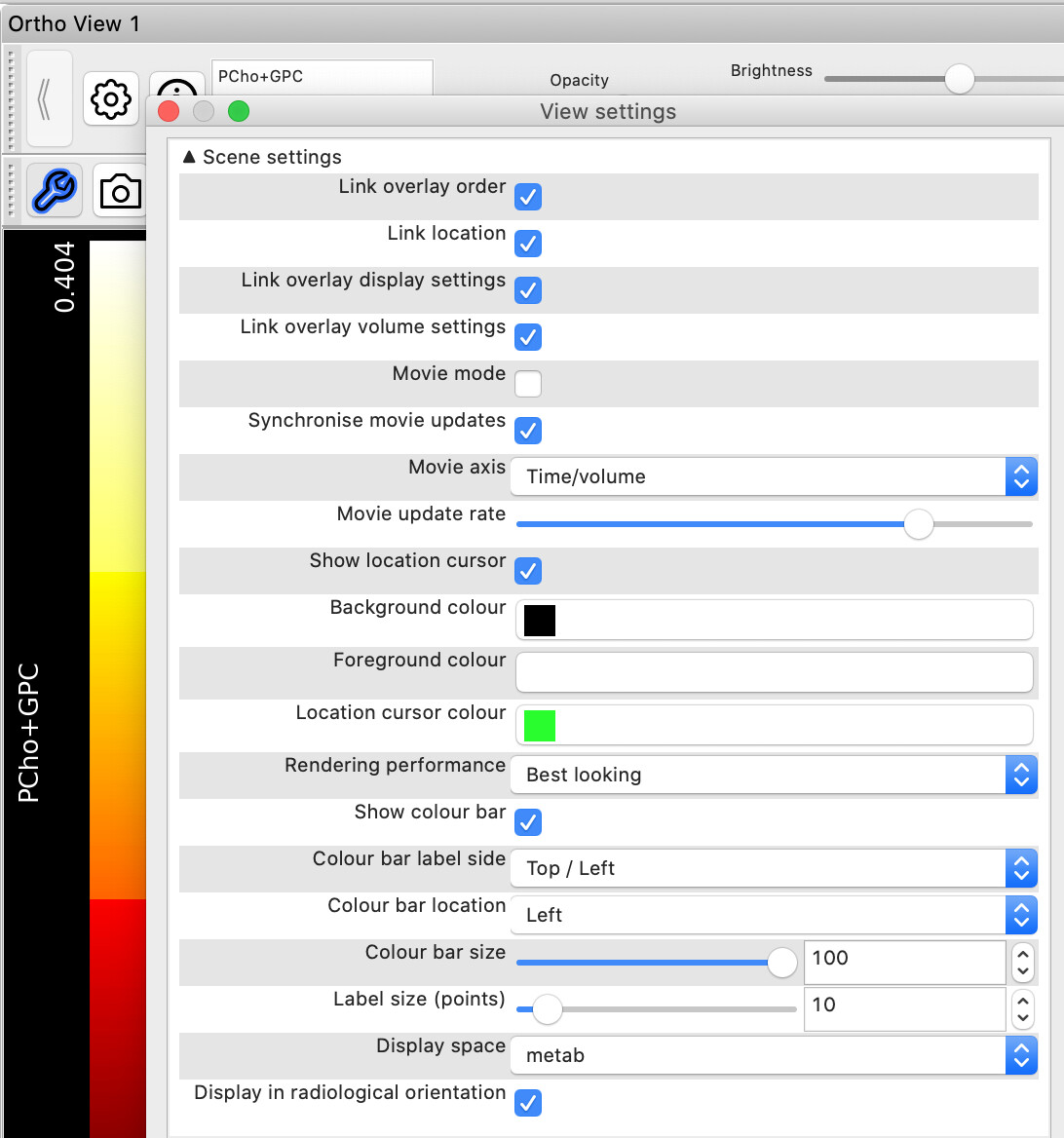
The last issue I guess is simply a display option which is dependent on the load order of the images into fsleyes interacting with an angled voxel. Once fsleyes is open try changing the Display space setting in the Otho panel View settings to the T1 or World Coordinates. This changes the base orientation axis of the whole ortho view.
Thanks for your quick reply! The orientation issue is solved!
I did create the data with spec2nii and it worked fine. The version I have is 0.3.4 I think it is the most recent?
I ran spec2nii philips . Can you clarify the spec2nii dump part?
For the segmentation part, we are hoping to not run any registration to standard space, are both FNRIT and FLIRT ran in svs_segment? Or is it common to be registering to MNI space for MRS?
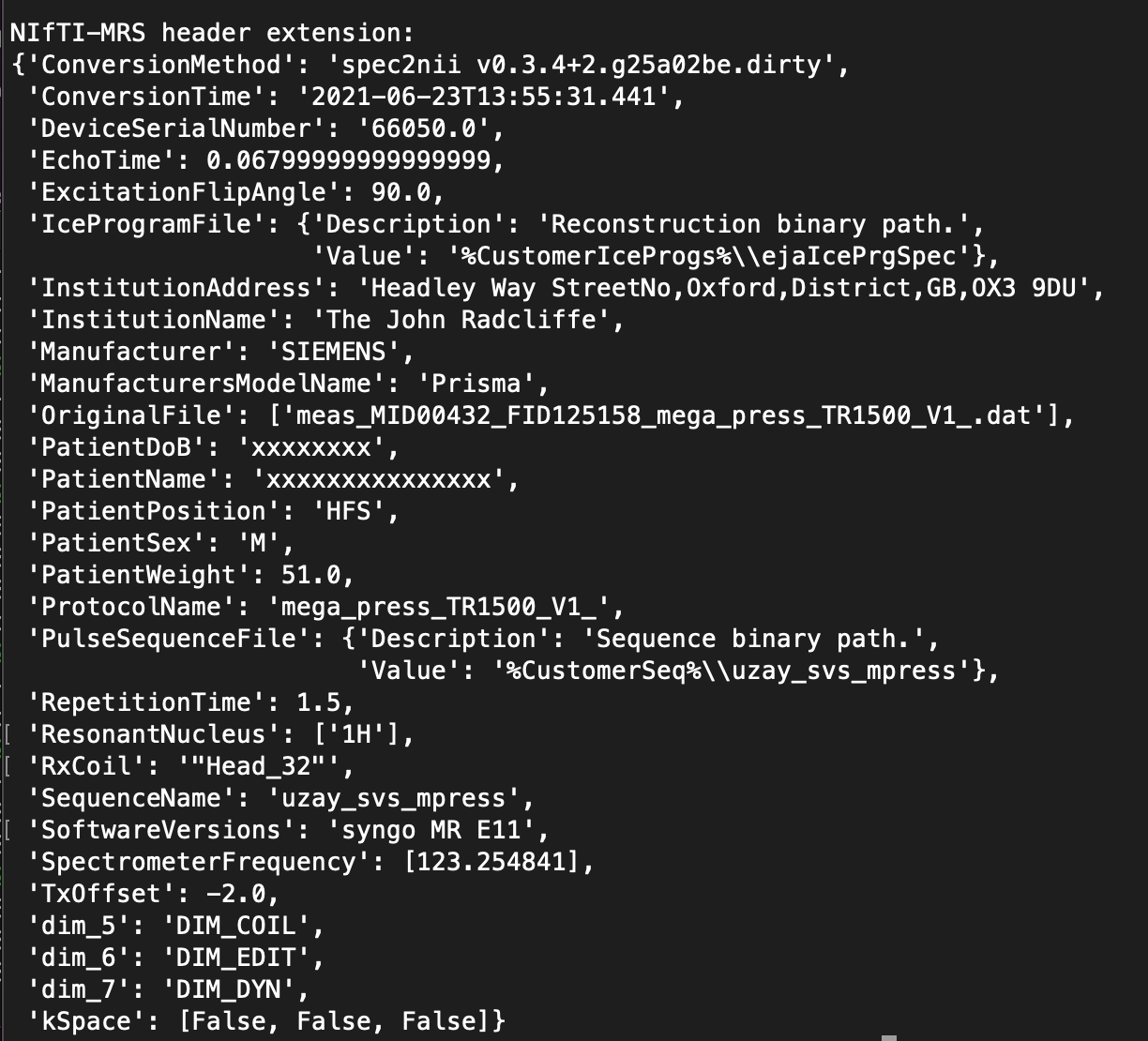
spec2nii dump myfile.nii.gz will print to stdout the Nibabel formatted NIfTI header and the NIfTI-MRS header extension (where all the MRS specific metadata is stored). The latter might look something like this
There are two required keys in this which are needed ‘SpectrometerFrequency’ and ‘ResonantNucleus’. In my example they are [123.25…] and [‘1H’]. Could you run the command on your data and post a picture here of what the ‘SpectrometerFrequency’ value is?
Looking closely at the error I think you might also have an old version of FSL-MRS (maybe one which comes with the main FSL installation). If you run fsl_mrs --version it should give you the version string. The most recent version is 1.1.2. I think you might have a version that was pre-NIfTI-MRS becoming the main file type (the problem with developing the data standard after the analysis program).
You can install the new version by following the instructions here.
Runs fsl_anat on the T1. fsl_anat does: fslreorient2std, robustfov, FAST bias field correction, registration to standard space (linear and non-linear) [FLIRT and FNIRT], brain-extraction [FNIRT-based or BET] and finally tissue-type segmentation [FAST].
Creates some 2x2x2 mock data with the same orientation affine as the svs voxel. Effectively a low resolution mask.
Uses FLIRT to register that mock data to the T1 to create a mask for the svs voxel in T1 space.
Masks and runs fslstats (mean) on the pve_n outputs of FAST.
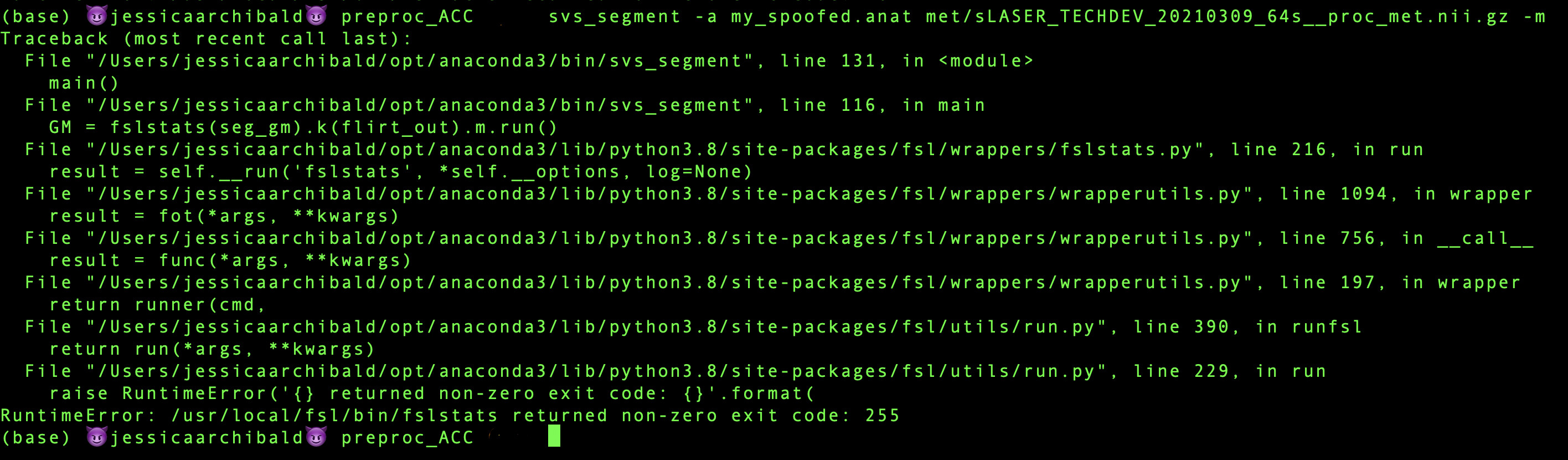
I think the messages you get about linear registration are coming from some of the steps run in fsl_anat. These steps aren’t used in the calculation of the FAST outputs. To check this you can spoof the fsl_anat step, replacing it with your own FAST output. Create a folder my_spoofed.anat and pass it to svs_segment as the -a option (also don’t include the -t option). In the folder you need a T1 called 'T1_biascorr.nii.gz' and the output of FAST 'T1_fast_pve_0.nii.gz', 'T1_fast_pve_1.nii.gz' and 'T1_fast_pve_2.nii.gz'. This will then not run the fsl_anat step and substitute your data into the calculation.
Of course you can also check what everything looks like in fsleyes. You can load the FAST results from both pipelines and compare. You can also load the mask created by svs_segment and verify that it matches the original svs voxel closely.
It was the version issue, sorry about that. I downloaded it right when it was announced in October last year and did not get around to trying it until now.
Thanks for this
I will go through the svs_segment info you shared and keep you posted.
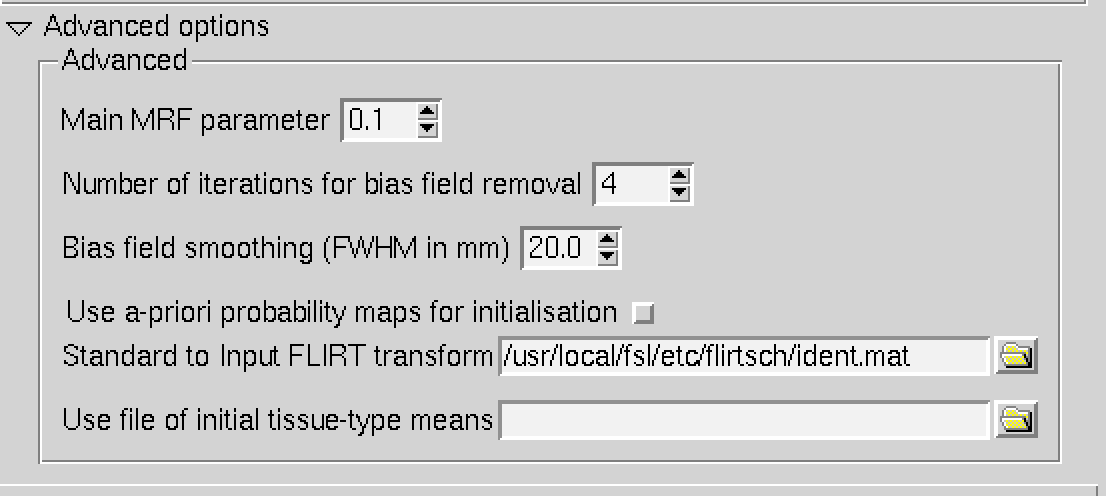
Thanks for explaining the svs_segment a bit more. I think the main difference is that I am running FAST with default settings not the bias field correction you listed above. I tried to run svs_segment with a spoofed anat folder with the 4 files you mentioned above. For the T1_biascorr I just gave it my 3DT1 but it gave an error :
it seems like I need a biascorr_brain_mask file… I did a bit of reading on FAST (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FAST) is the bias field correction you mentioned in the advanced options?
Sorry, I missed this. The forum used to notify me of replies, probably something I changed.
It’s a good question. I use fsl_anat - FslWiki which interleaves a number of FSL steps. Looking at the source code for fsl_anat there isn’t a simple correspondence between what it does and the FAST options above. I think fsl_anat is now the recommended option for this sort of stuff, but it’s important we get to match everything up first with your FAST results.
I don’t do anything particularly clever with the brain mask, its just in there to check that the voxel actually fully intersects with the area identified as brain. You could take a mask generated by FSL bet and rename it appropriately i.e. to biascorr_brain_mask, and place it in your spoofed anat folder. That should then run.
I’ll add this to the list of things to make a bit easier.
Thanks for your follow-up on this! I was able to get the mask to be made by replacing the biascorr_brain_mask with the BET file. When I compare the location to our voxel it looks great. However, the segmentation step failed.
From what I can see fslstats is using some output from flirt? I will spend a bit more time looking at the fsl_anat Wiki you sent above. Thanks again for your help on this and I will keep you posted
Good that the voxels line up. That error isn’t that useful to diagnose the issue, but it looks like it is probably that one of the two inputs is missing.
On line 116 it runs the equivalent of fslstats -t /fast_output/fast_output_pve_1 -k SVS_mask.nii –m, it does this using the fslpy interface so the call looks like fslstats(seg_gm).k(flirt_out).m.run().
If it is a missing file, it’s unlikely to be the mask file as the exact same command has just been run for the csf fraction on line 115. seg_gm is where I would look that is looking for the file my_spoofed.anat/T1_fast_pve_1.nii.gz. Does that exist?
If you want to have a look at the errors in a debugger a trick is to run
python -m pdb -c continue ‘which svs_segment’ -a my_spoofed.anat path/to/mrs.niii.gz
you can then use the pdb commands to inspect any of the variables. Note those quotes around which svs_segment should be backquotes but I can’t figure out how to escape backquotes in the forum.
 )but when running the mrs_vis command I get this error:
)but when running the mrs_vis command I get this error: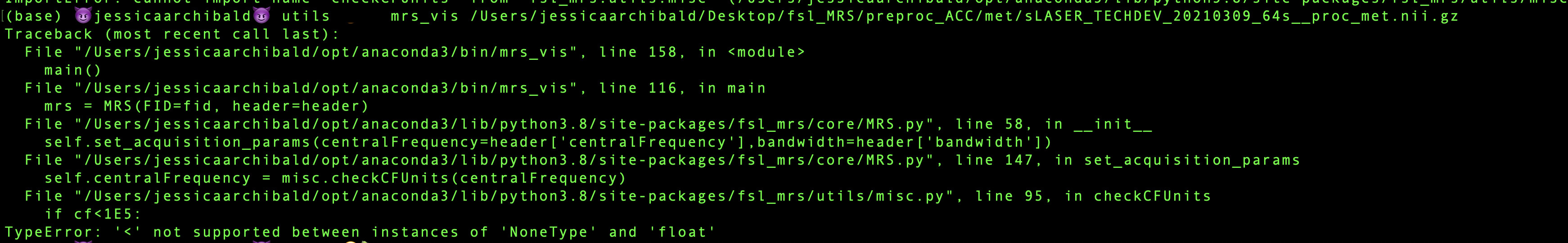
 . The file is fine, when I visualize it in MRIcroGL or Matlab, but just curious if other are running into this issue as well. This occurs when I load the unsegmented 3DT1 as well as the segmented and betted files.
. The file is fine, when I visualize it in MRIcroGL or Matlab, but just curious if other are running into this issue as well. This occurs when I load the unsegmented 3DT1 as well as the segmented and betted files.