Hello everyone,
first of all thank you @alex and @wclarke for the detailed information.
As you suggested, I did some preprocessing steps with the fsl_mrs subcommands. The metab data looked nice unprocessed, but the water reference looked not fully processed when I visualized it.
For example the right putamen:
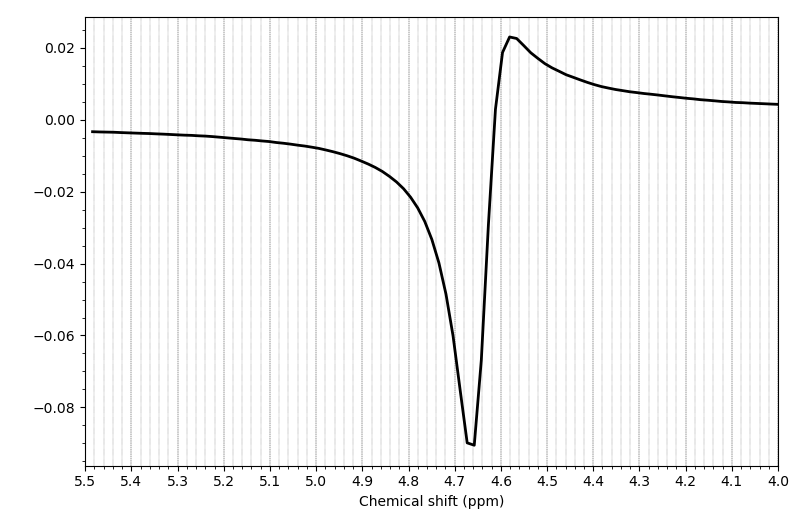
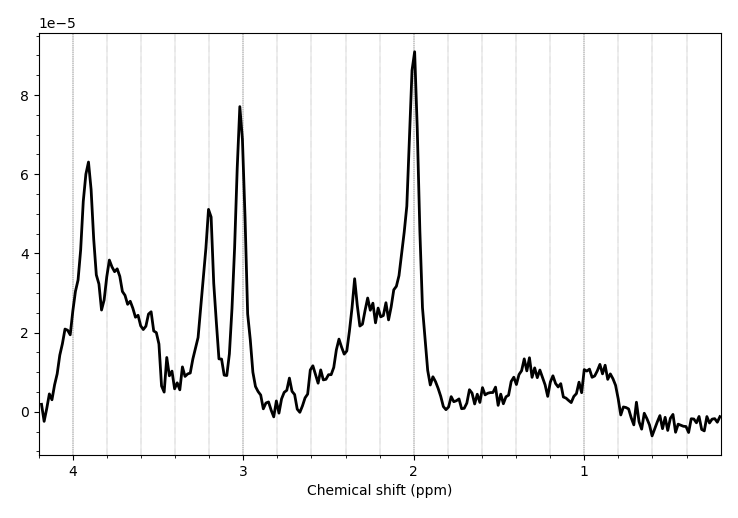
So I tried performing the remaining preprocessing steps. I used the following steps:
Eddy current correction (ECC) for both (metab and ref).
Centering the echo as described in the tutorial, although I wasn’t sure if these were the right parameters for my sequence (fsl_mrs_proc truncate --file fsl_mrs_proc/file_ecc.nii.gz --points -1 --pos first --filename file_trunc --output fsl_mrs_proc -r)
Removal of residual water
Phase correction
After ECC, the visualization of my metabolites looked a bit weird, but the reference file was fine. When visualizing the final processed files, the metab.nii looked better than after ECC, but seemed a bit shifted in one example. So I’m not sure if all the steps were required or even if I left out an important step.
(This example is the acc from a different subject than the one whose folder I had provided you.)
Before processing:
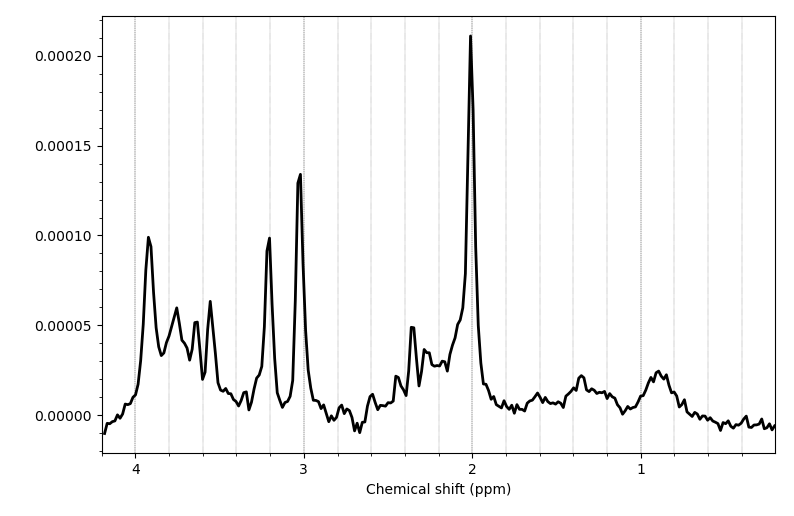
After processing:
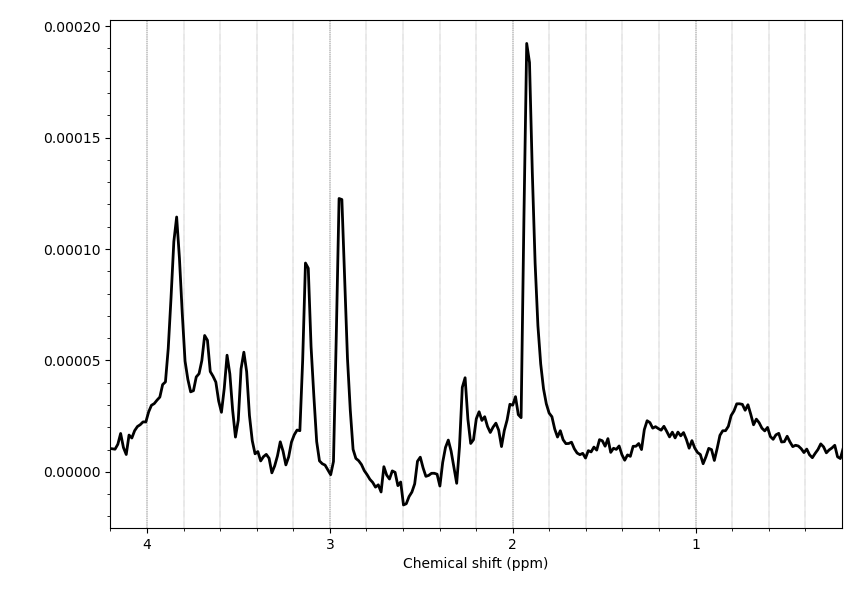
The processed file to the examples before (putamen on the right), however, looks good.

What is your opinion, should I do the preprocessing steps?
The next problem occurred when I tried to fit my data. As I understood it, I can’t use the basis spectra files from the example in the fsl_mrs tutorial, because they were created for the STEAM sequence.
So, first I tried to create my own basis spectra using the fls_sim function, but unfortunately I wasn’t able to create a json file as I couldn’t figure out all sequence description parameters and sequence block parameters which were described on https://open.win.ox.ac.uk/pages/fsl/fsl_mrs/seq_file_params.html#seq-file-params.
So I tried to find a basis spectra file for a philips PRESS 35 sequence.
I found one .basis file at this page (http://s-provencher.com/lcm-basis.shtml) but when I wanted to apply it, I got this back:
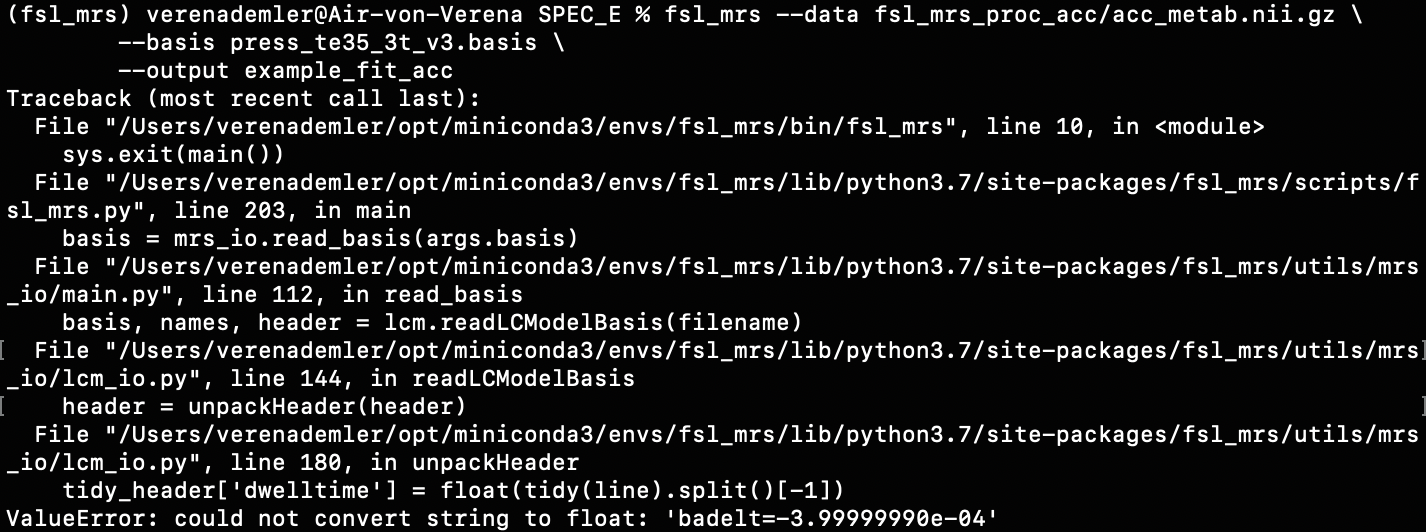
Then I found a folder (on MR Spectroscopy Basis Sets | MR SCIENCE Laboratory) that contained .raw files for the various metabols, but unfortunately that didn’t work either.
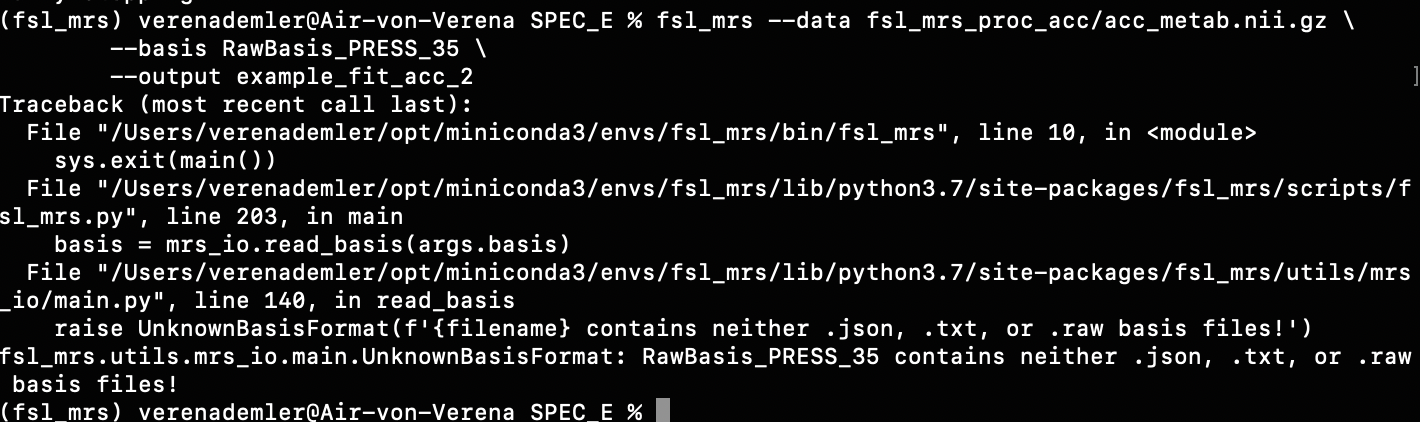
Do you have any idea how I could solve this problem?
I am sorry for asking so many questions, but unfortunately I have no more ideas of my own what I could try. So, I would be very grateful if you could help me.
If you need more information, here is a description of my acc sequence and otherwise feel free to ask me.
ACC_SV_PRESS_35.txt (2.6 KB)
Many thanks,
Verena