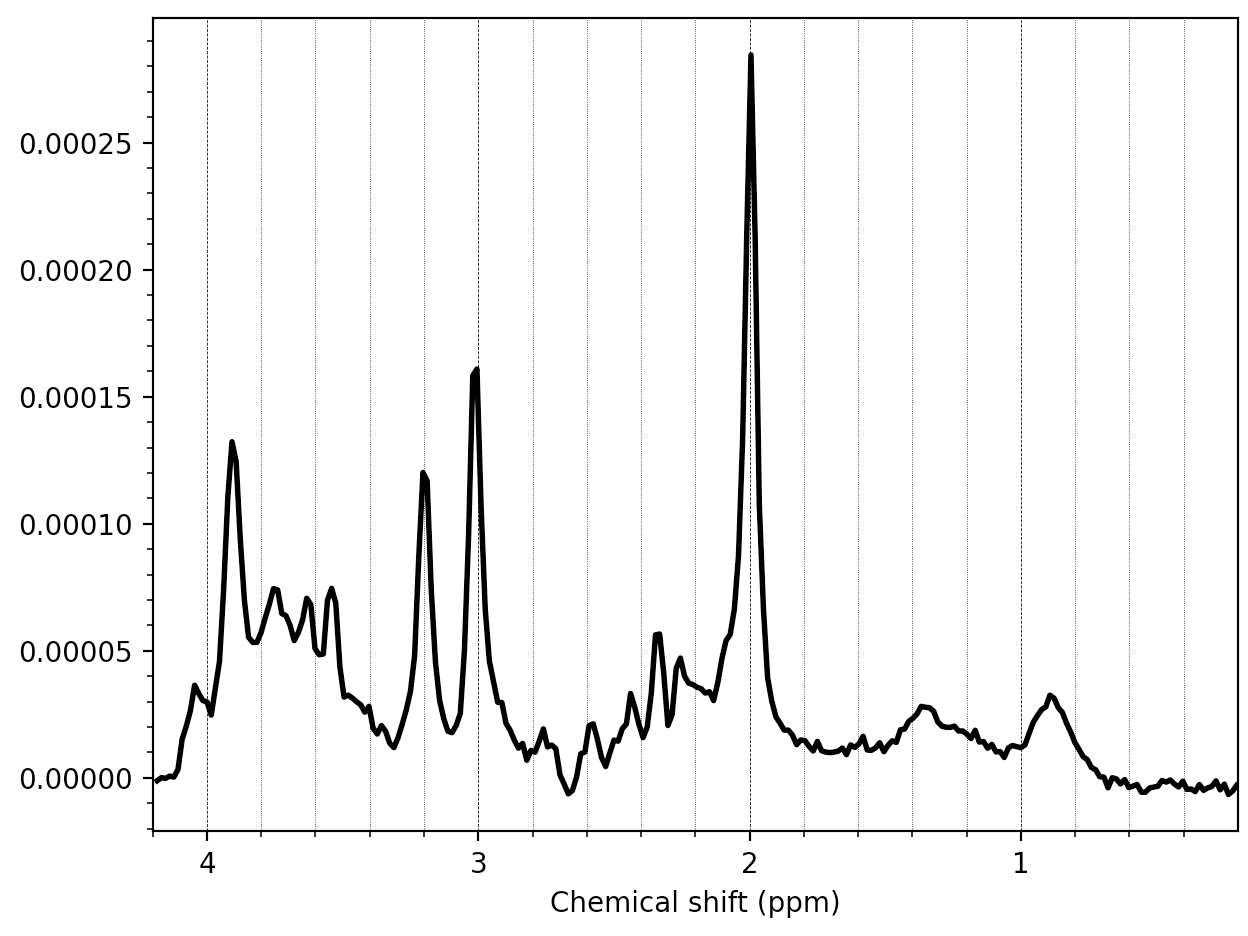
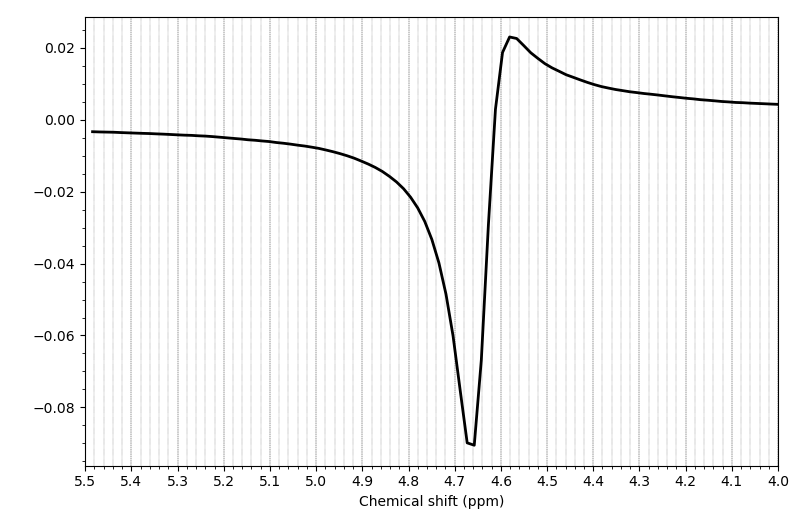
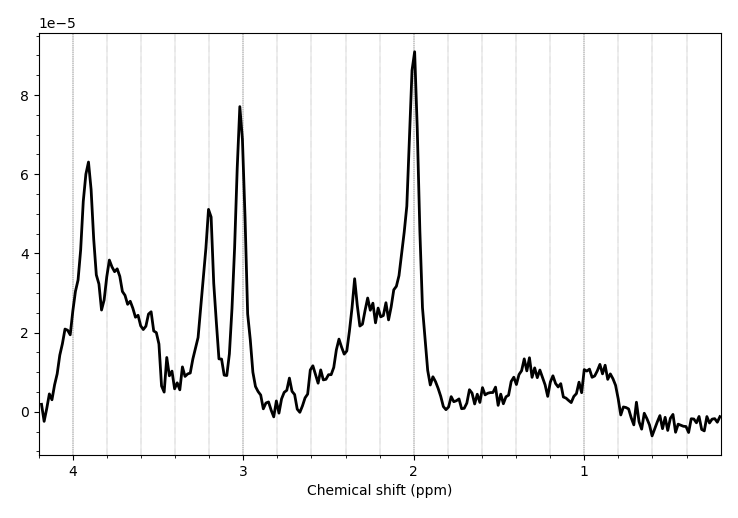
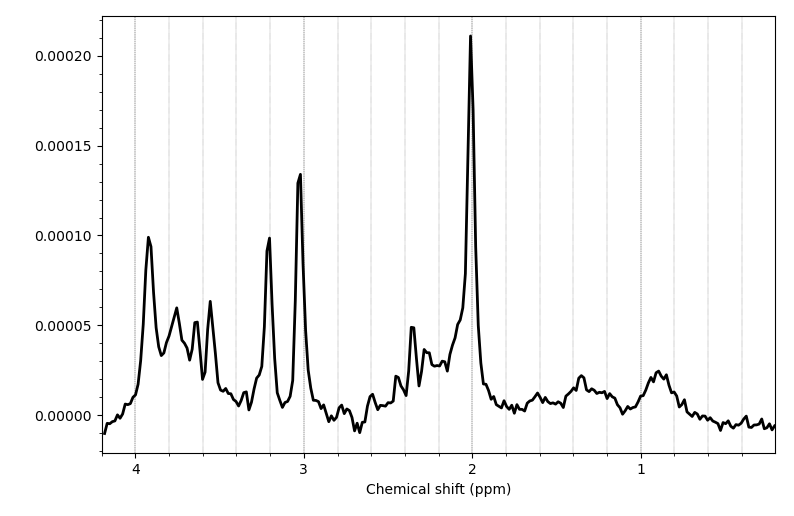
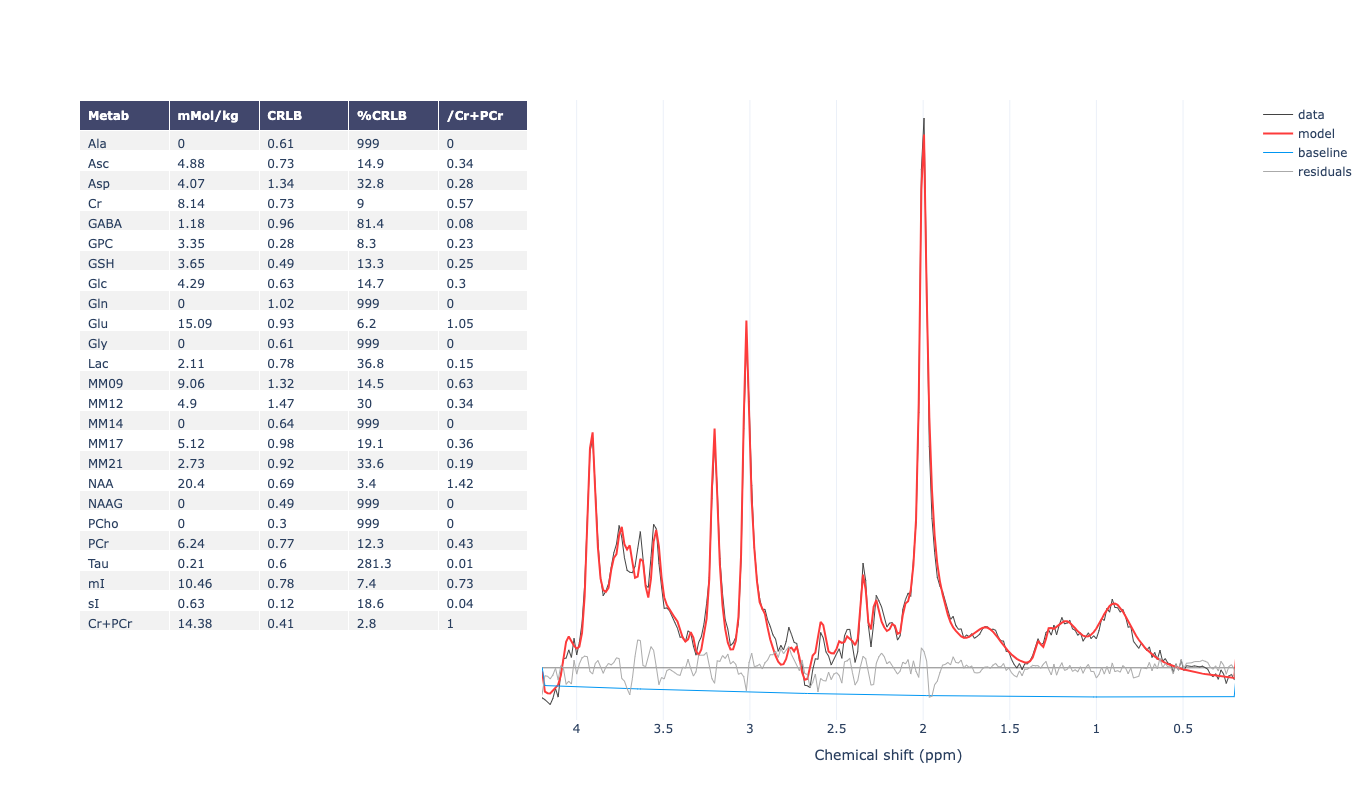
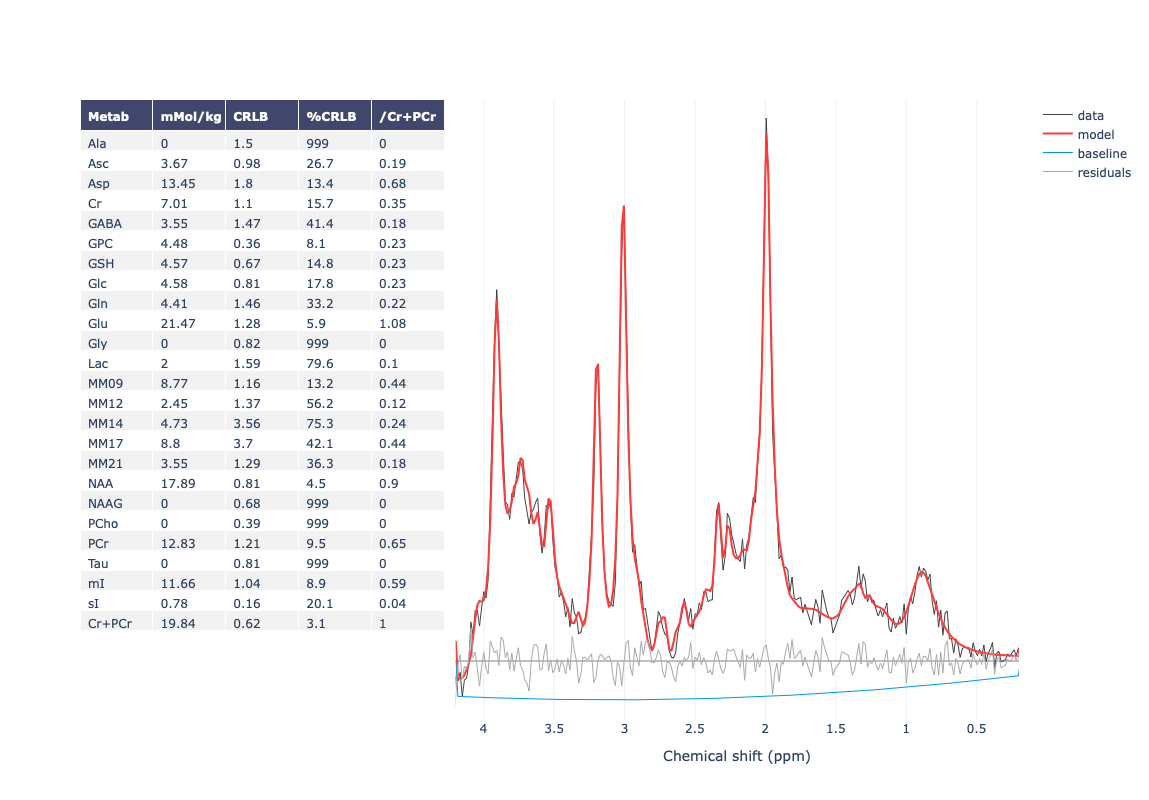
Hello everyone,
I’m a medical doctorate in a Neuropsychiatry and Neuroimaging Lab and we just begun to use FSL-MRS (version: 1.1.13) for our spectroscopy data.
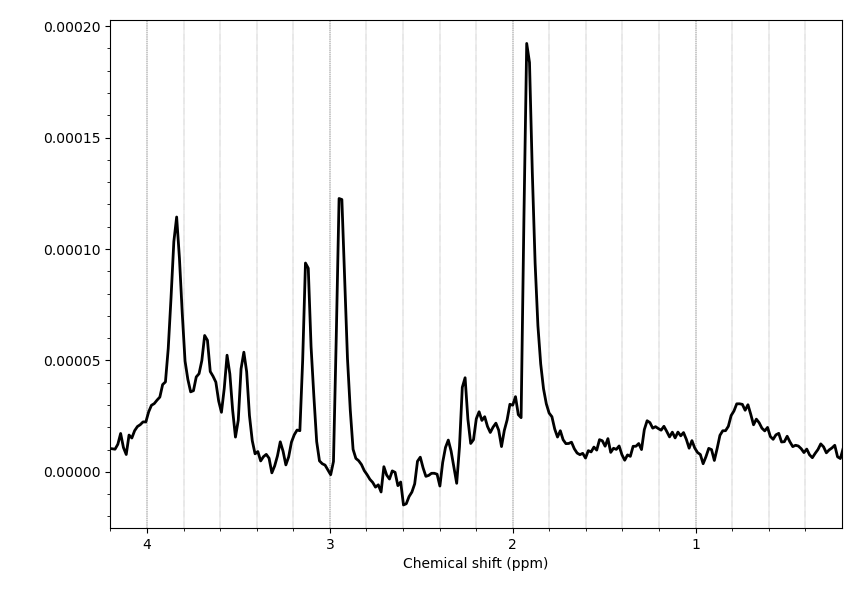
We acquired our data in a Philips Ingenia Elition 3.0T X and saved them as enhanced dicom files. The scanner produced two files one raw dicom and one spectroscopy file. I only used the second one for the further steps. Converting these files with spec2nii philips_dcm works, but I am having some issues with the dimensions and the following processing steps.
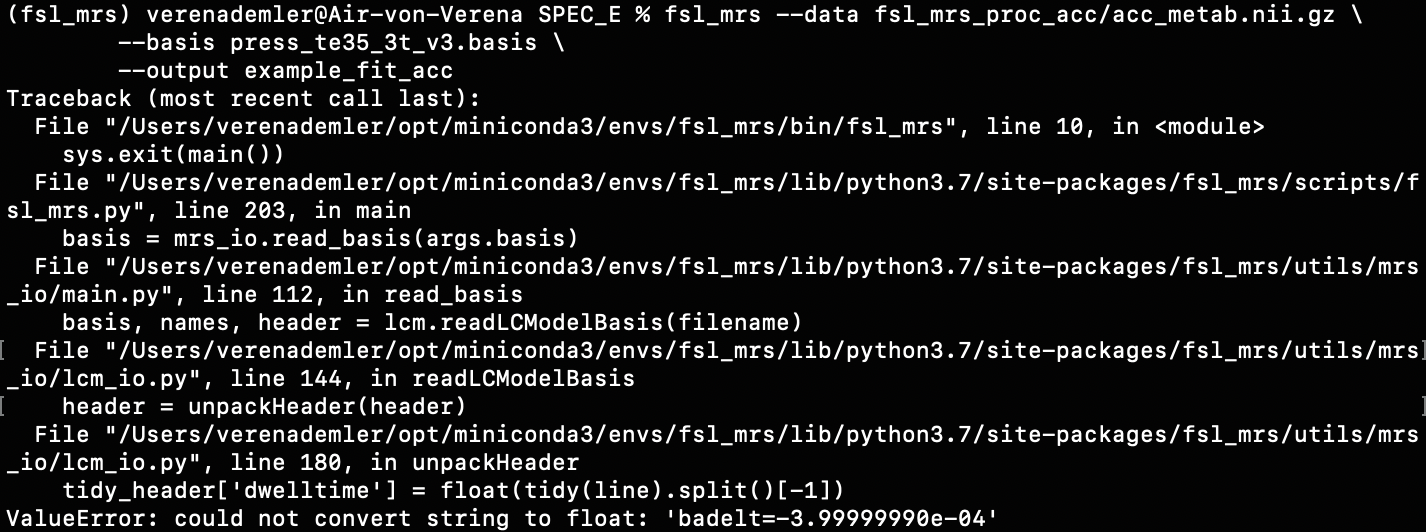
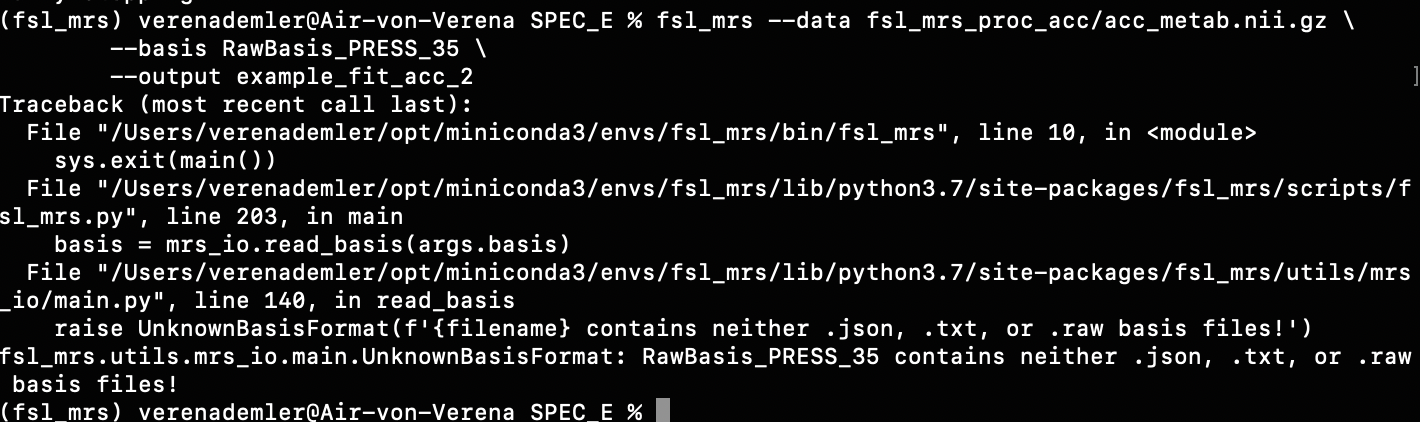
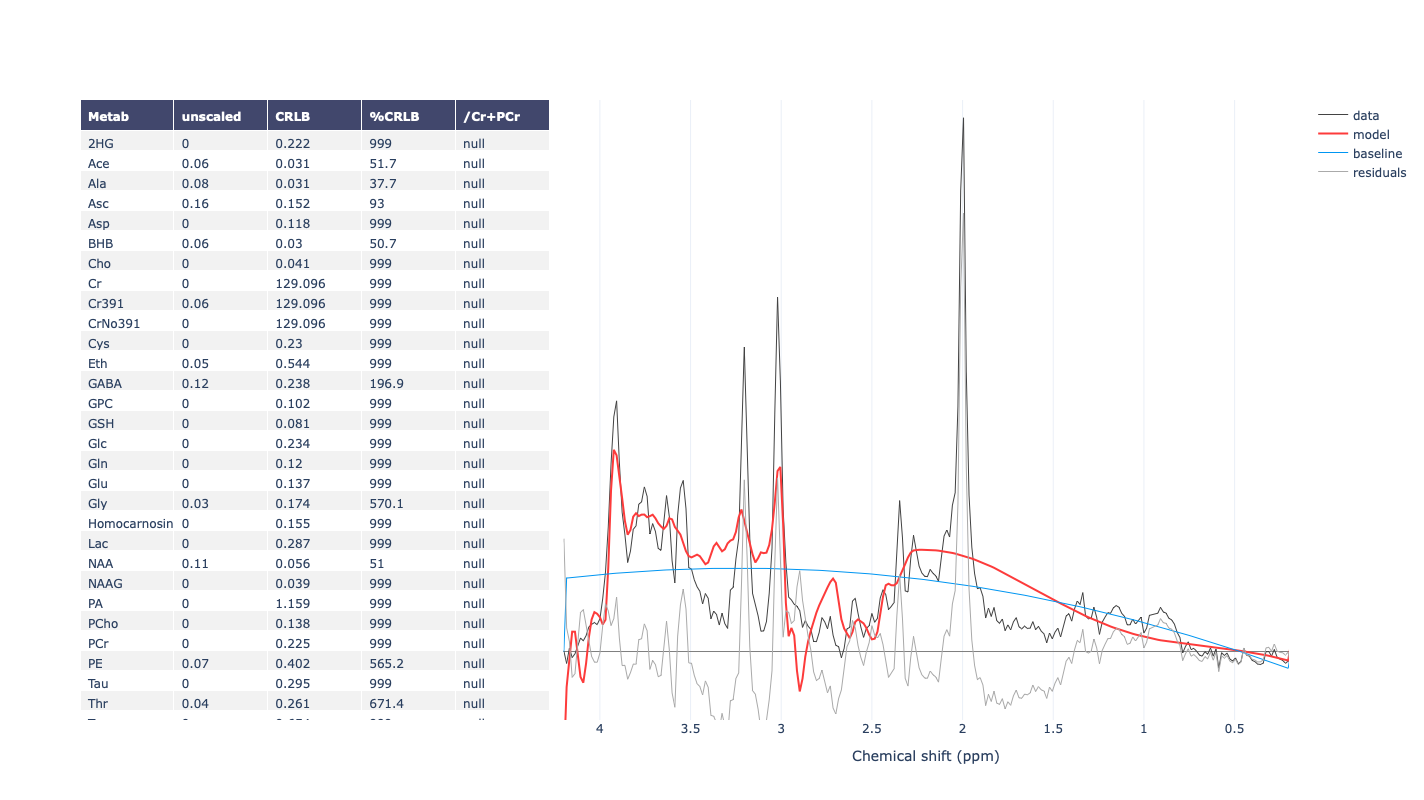
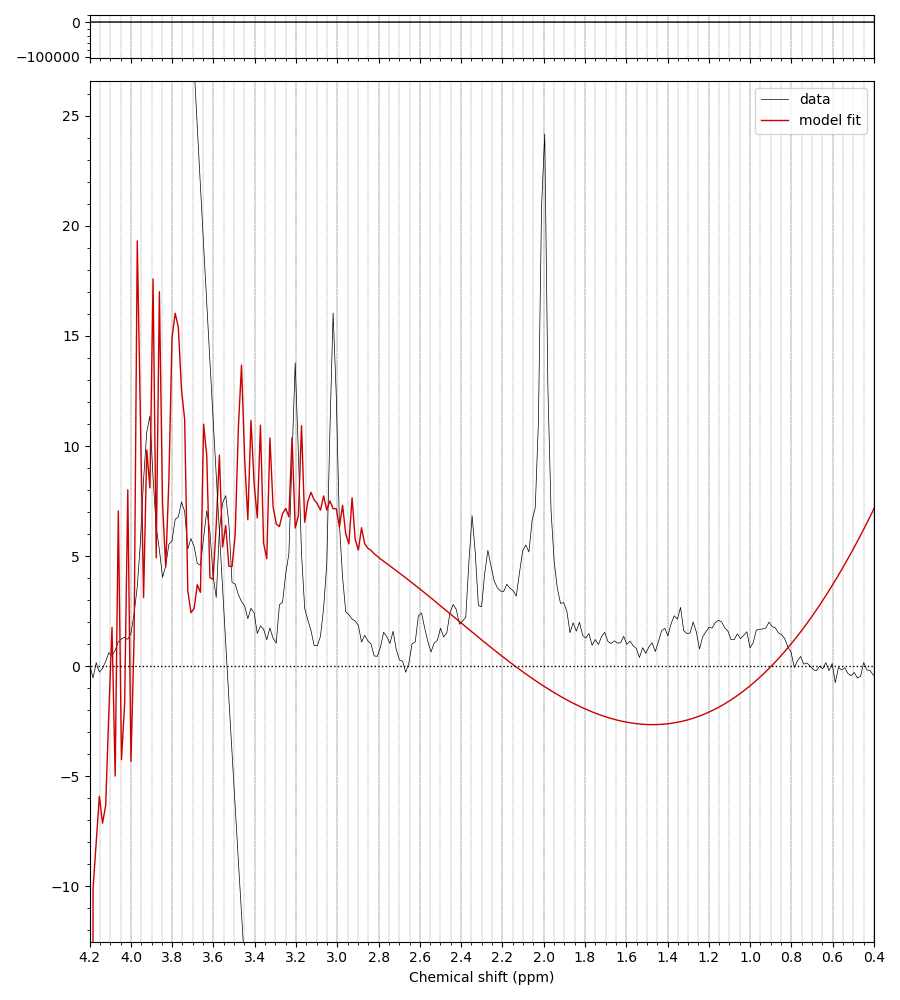
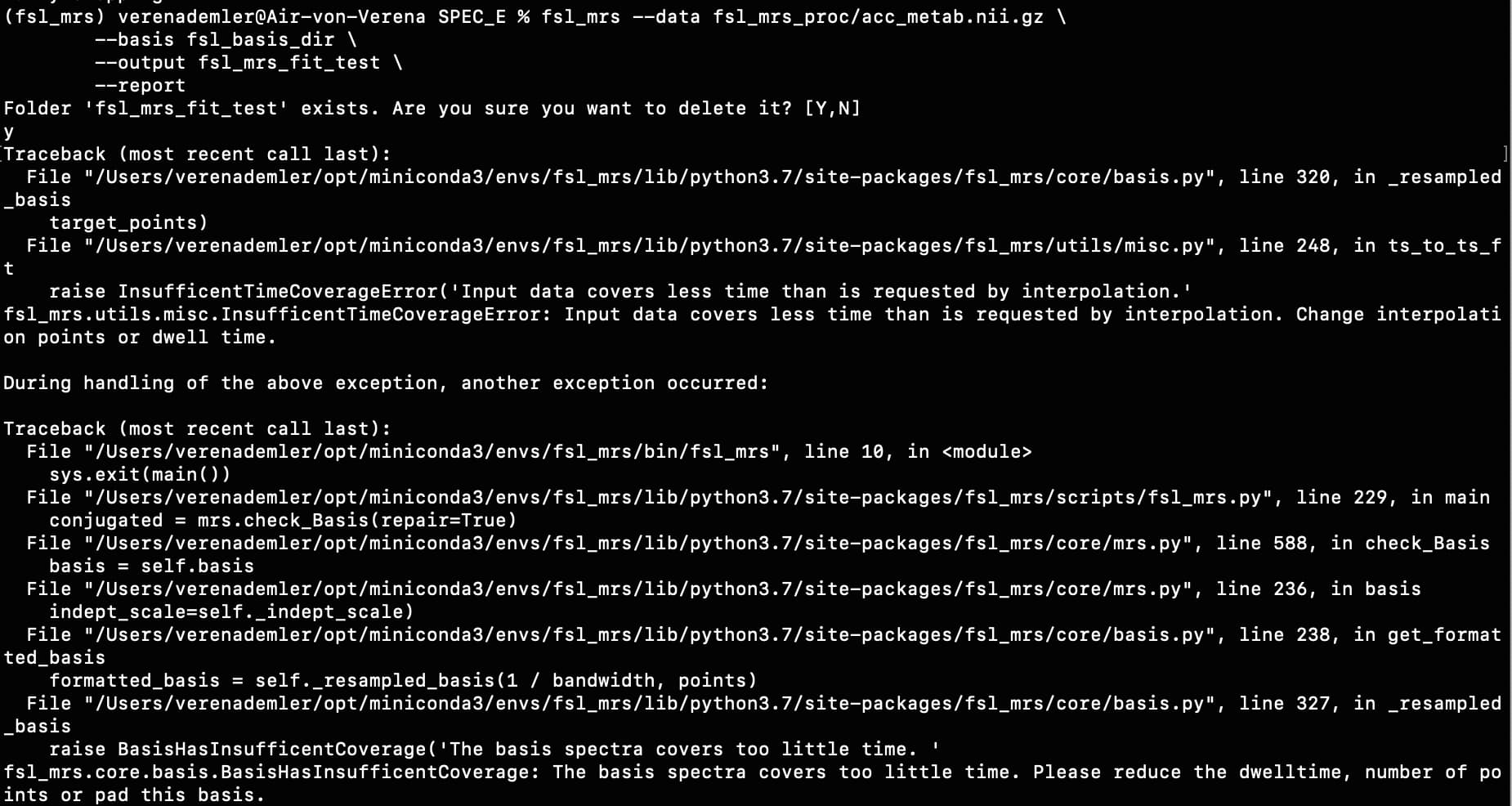
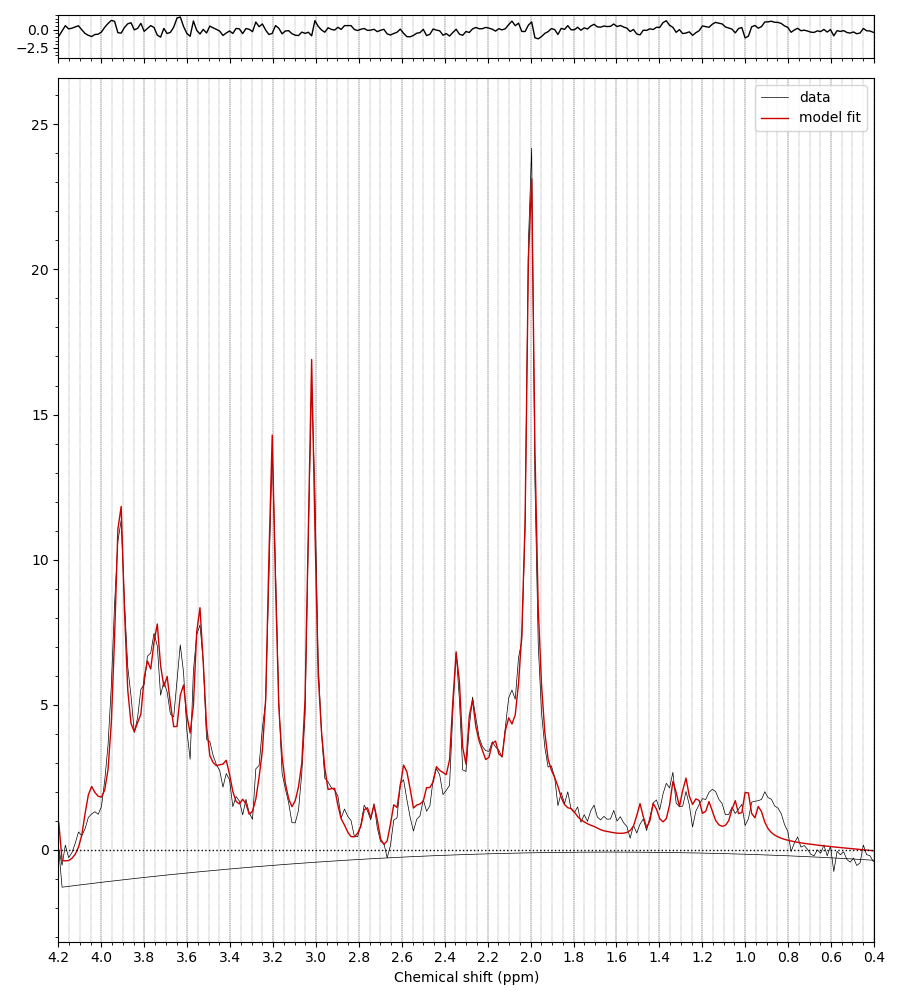
For example, when I run the predefined fsl_mrs preproc ( fsl_mrs_preproc --output processed --data file.nii.gz --reference file_ref.nii.gz --report ) I’ m getting this error:
ValueError: remove_unlike only makes sense for data with a dynamic dimension
And when I use mrs_tools info to see the dimensionality of the data, the 5th and 6th dimensions are not specified.
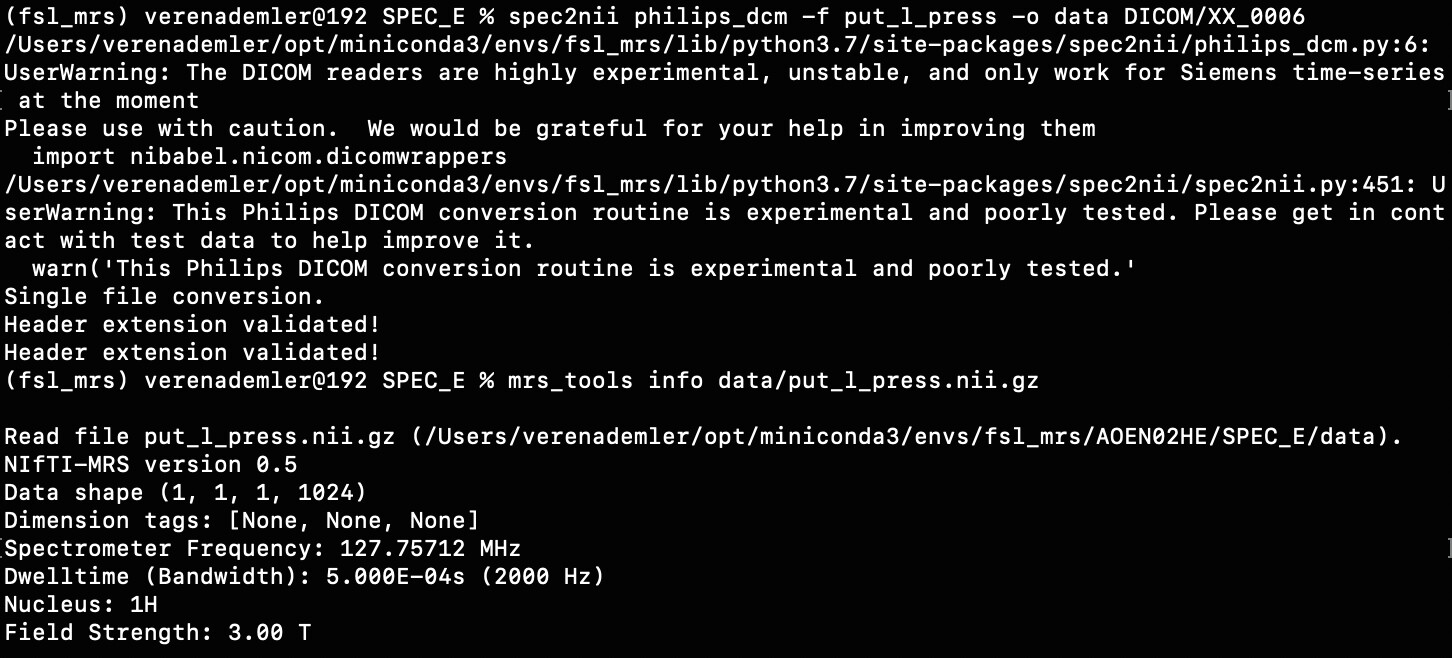
So, now for my questions:
Did I make a mistake converting my files from Dicom to Nifti? Are these dimensions essential or can I change or omit the preprocessing steps that use these dimensions? Or is there any other way?
I have attached a folder with the enhanced dicom files as well as the output from spec2nii dump.
Please let me know if you would like any additional information.
Many thanks,
Verena
spec2nii_dump.txt (2.2 KB)
SPEC_data.zip (733.6 KB)