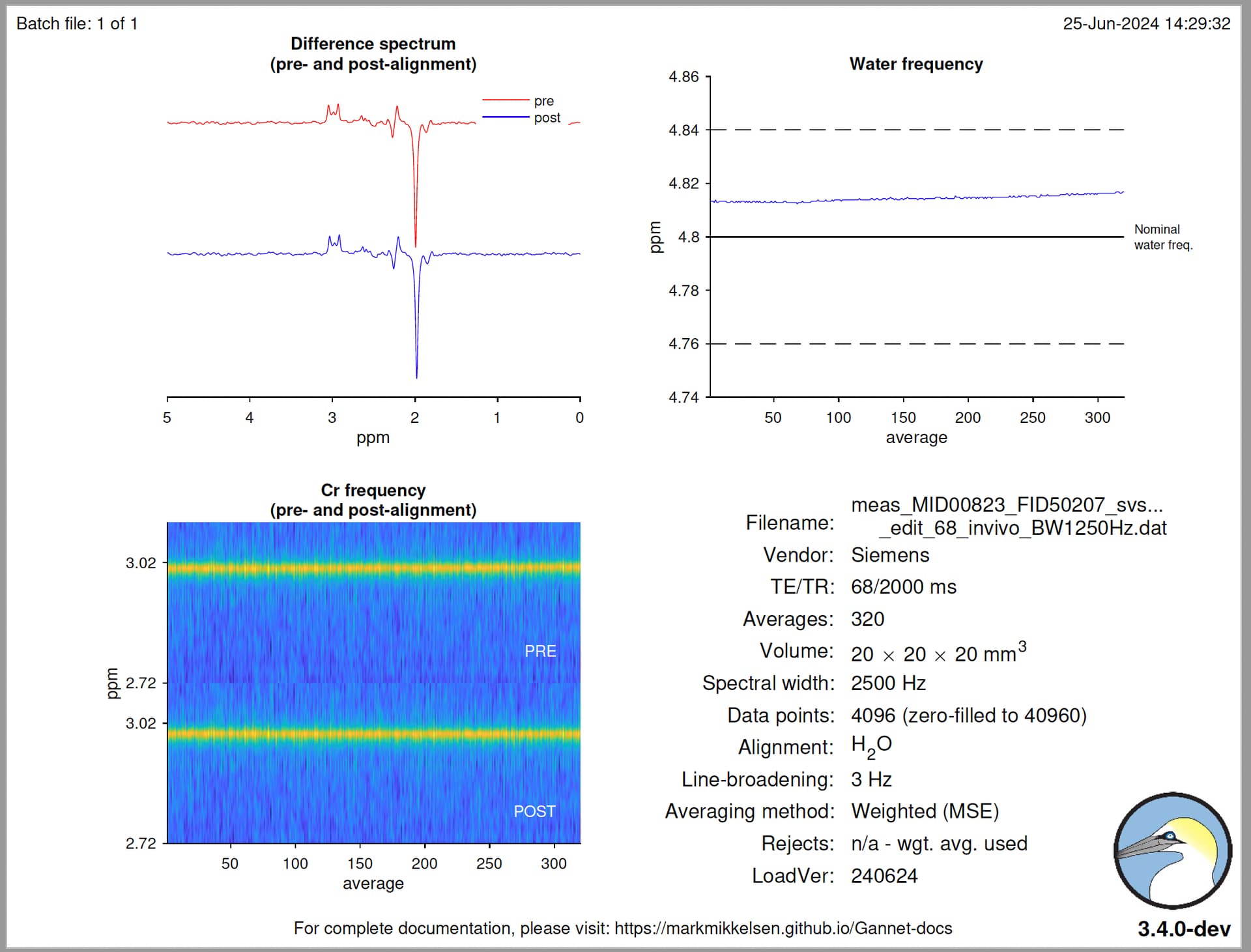
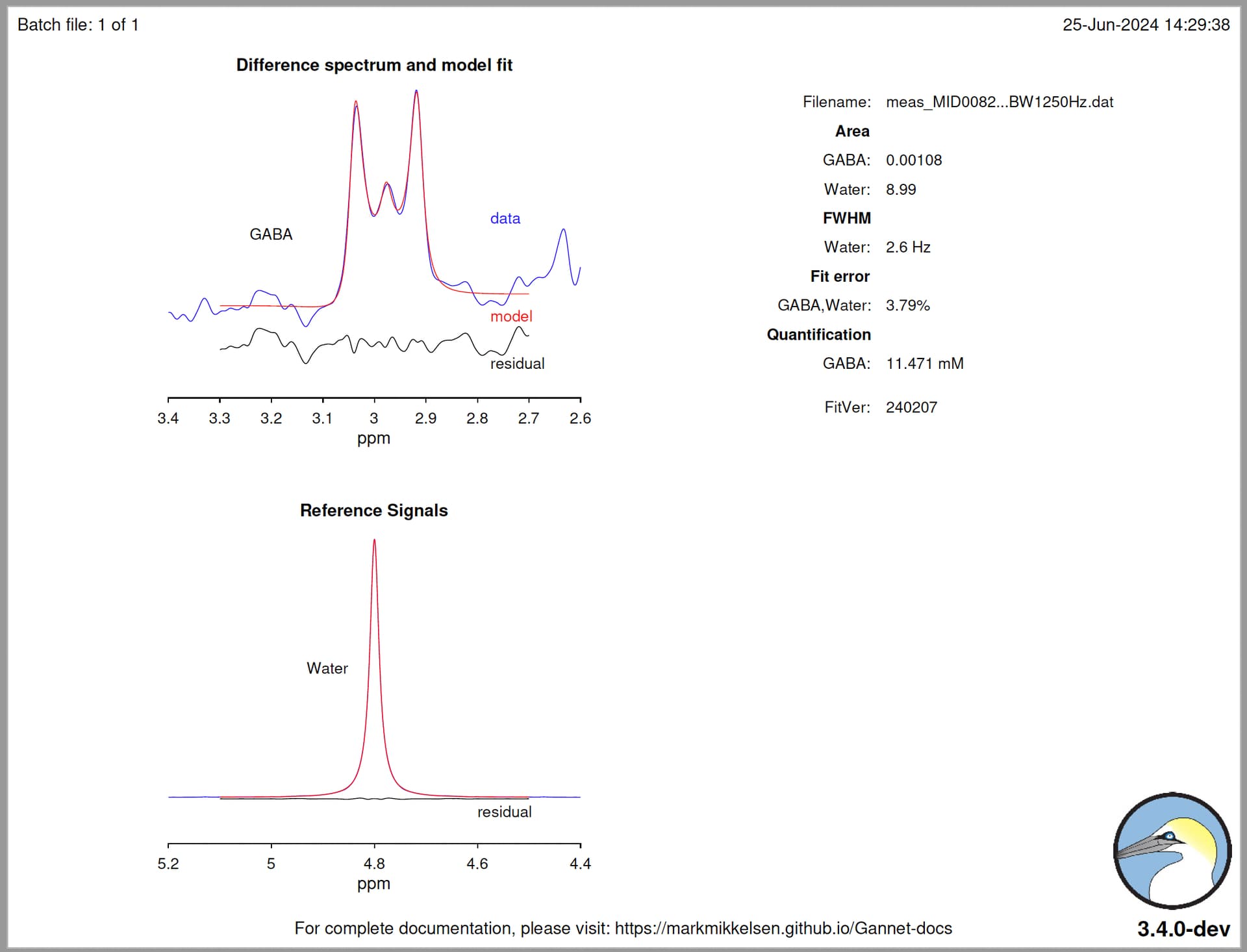
Hello @mmikkel,
Sorry for the delay and thanks for the reply 
I had understood that .dat files were just water data, so I was trying to use .IMA files. But any of them are interesting to me.
I downloaded the development version of Gannet (v3.4.0-dev), but I still couldn’t open the data using GannetLoad(). It appears that the code gets stuck in a loop when the function twix_obj = mapVBVD_Gannet(fname); is called on line 193 by SiemensTWIXRead().
This is my command line:
MRS = GannetLoad({'/home/wesna/MRS/Phantom/data-megapress-spectra/E1/RAW/GABA11_NAA_15mM_Cr_8mM/meas_MID00823_FID50207_svs_edit_68_invivo_BW1250Hz.dat'}, {'/home/wesna/MRS/Phantom/data-megapress-spectra/E1/RAW/GABA11_NAA_15mM_Cr_8mM/meas_MID00819_FID50203_svs_se_68_WS_OFF.dat'});
And my configuration:
function MRS_struct = GannetPreInitialise(MRS_struct)
% Acquisition parameters
MRS_struct.p.target = {'GABA'}; % Edited metabolite(s) of interest; permitted options are:
% If MEGA-PRESS:
% {'GABA'}, {'GABAGlx'}, {'GSH'}, {'Lac'}, or {'EtOH'}
% If HERMES:
% {'GABAGlx','GSH'}, {'Lac','GSH'}, or {'EtOH','GABA','GSH'}
% If HERCULES:
% {'GABAGlx','GSH'}
% If phantom data:
% and MEGA-PRESS: {'GABA'}, {'Glx'}, {'GSH'}, {'Lac'}, or {'EtOH'}
% and HERMES: {'GABA','GSH'}, {'Glx','GSH'}, {'Lac','GSH'}, or {'EtOH','GABA','GSH'}
MRS_struct.p.ON_OFF_order = 'onfirst'; % The editing order applied at acquisition.
% If empty (the default: []), Gannet will determine the editing order automatically.
% Otherwise, input 'offfirst' or 'onfirst' for MEGA-edited data;
% or, for HERMES/HERCULES data, input a four-letter combination, such as 'ABCD' or 'CBAD', etc.
% (see: doi:10.1016/j.neuroimage.2016.07.056)
MRS_struct.p.seqorig = 'JHU'; % Origin of Philips MEGA-PRESS or GE HERMES sequences;
% options are 'JHU' or 'Philips' if Philips, or 'Lythgoe' if GE (for HERMES only)
% Analysis parameters
MRS_struct.p.LB = 3; % Exponential line-broadening (in Hz); default = 3 Hz; for phantom data, ~1.5 Hz is recommended
MRS_struct.p.water_ECC = 1; % 1 = YES, perform eddy current correction on water data
MRS_struct.p.metab_ECC = 0; % 1 = YES, perform eddy current correction on metabolite data (requires a water reference)
MRS_struct.p.water_removal = 1; % 1 = YES, remove residual water signal in the DIFF spectrum using HSVD
MRS_struct.p.alignment = 'H2O'; % Alignment method; options are 'RobustSpecReg' (recommended), 'SpecReg', 'SpecRegHERMES',
% 'Cr', 'Cho', 'NAA', 'H2O', or 'none' (recommended for phantom data)
MRS_struct.p.use_prealign_ref = 0; % 1 = YES; in some cases, using RobustSpecReg to align HERMES/HERCULES data can result in
% worse alignment compared to the pre-aligned data; setting this parameter to 1 will
% make RobustSpecReg use the averaged pre-aligned subspectra as references to align the
% averaged post-aligned subspectra, which may improve the final alignment
MRS_struct.p.vox = {'vox1'}; % For naming voxels, e.g., {'DLPFC'}; if data were acquired using PRIAM this could be,
% e.g., {'anterior','posterior'}, {'right','left'}, etc.
MRS_struct.p.fit_resid_water = 0; % 1 = YES, fit the residual water signal in the OFF spectrum to calculate a water suppression factor
MRS_struct.p.weighted_averaging = 1; % 1 = YES, average subspectra using weighted averaging; otherwise, use arithmetic averaging
% Flags(0 = NO; 1 = YES)
MRS_struct.p.HERMES = 0; % Data were acquired using HERMES
MRS_struct.p.HERCULES = 0; % Data were acquired using HERCULES; if 1, MRS_struct.p.HERMES must be set to 1 as well
MRS_struct.p.PRIAM = 0; % Data were acquired using PRIAM
MRS_struct.p.phantom = 1; % Data are from a phantom (assumes phantom was scanned at room temperature)
MRS_struct.p.join = 0; % Join multiple files (this can be performed in batch to join files across multiple subjects)
MRS_struct.p.mat = 0; % Save MRS_struct as a .mat file
MRS_struct.p.csv = 0; % Extract useful results from the output structure MRS_struct and export them to a .csv file (applies to
% GannetFit, GannetSegment, and GannetQuantify only)
MRS_struct.p.normalize = 0; % If 1, the voxel masks created by GannetCoRegister and GannetSegment are normalized to MNI space
% and, if more than dataset has been run in the pipeline, a mean overlap voxel is created
% (note that this is only run if GannetSegment is run)
MRS_struct.p.append = 0; % Append PDF outputs into one PDF (separately for each module) (requires export_fig in the Gannet
% directory to be added to the search path and Ghostscript to be installed)
MRS_struct.p.hide = 0; % Do not display output figures
end
Any ideas what could be wrong? Did you just change the version and the code worked? I also tried passing just the file name as an argument, but the code remains stuck in the loop (the code has no errors, but continues to load infinitely).
Thank you!