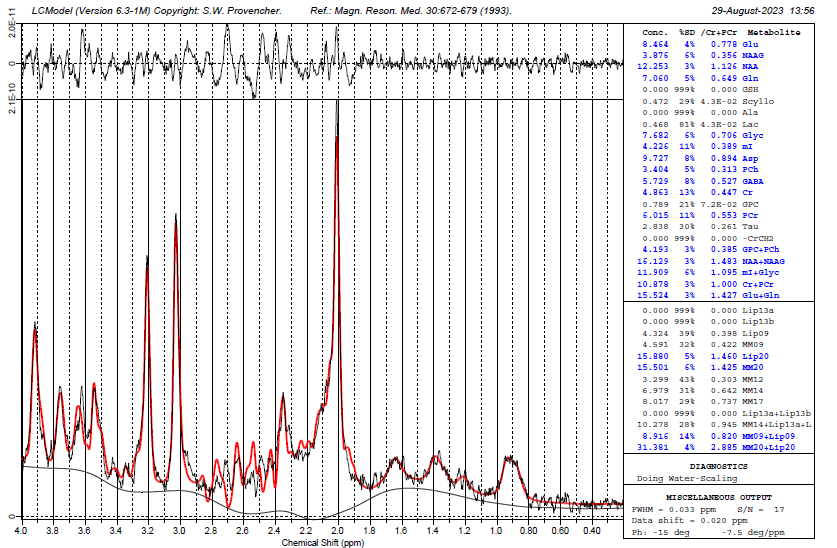
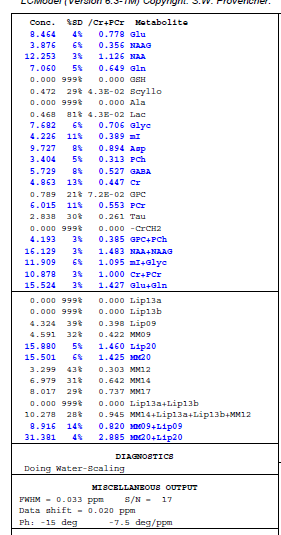
I’m having a super frustrating time being able to quantify GSH from data that we have. I don’t really know where the issue is. It is data from a 7T siemens machine using sLASER sequence. After running through our processing pipeline I’m getting 0 for nearly all the participants and that just doesn’t seem that it should be the case. Feels unlikely it’s all bad GSH data…but maybe I’m wrong. Oddly, I noticed that someone had accidently used the steam basis set we have to process the sLASER data and it fits GSH… does this mean it’s a problem with our sLASER basis set??
Sorry if none of that makes sense… I’m learning and troubleshooting at the same time.
To my eye, that basis set is not a good fit for the data. The NAA multiplet peaks are very poorly modelled, the GABA/Glu peak around 2.3 ppm is off and there are issues around 3.65 ppm as well.
What’s the precise sequence? Is it one of the CMRR sequences, if so which one? Unfortunately “sLASER” + a single TE isn’t enough to characterise the sequence well enough to generate a good basis set. If you can provide the protocol PDF it’s possible to come up with a very accurate sequence description that can be fed into either MARSS or FSL.
First, let me thank you both @admin and @wclarke for being so so helpful, knowledgeable, and responsive!!! It is so great to have this community!!!
It is indeed a CMRR sequence. I don’t know much of anything about generating a basis set but I did find the MARSS file and could look inside for what sequence info was used when it was generated. But I do have a protocol pdf that I can send separately!
You might also ask the CMRR folks who gave you the sequence for a matching basis set. If you don’t know where exactly it’s from, you should be able to peek into the data headers to find a sequence string that identifies the author and either starts with eja (Eddy Auerbach) or dkd (Dinesh Deelchand).
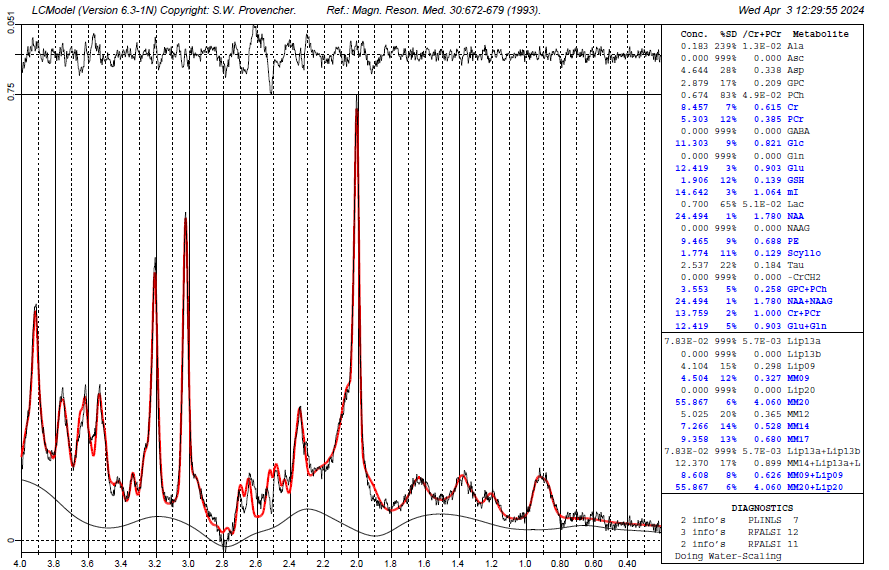
Revisiting here with an update…I’m getting a fit now for GSH thanks to the wonderful resources from @wclarke code for the CMRR slaser 7T basis set simulations.
I am still struggling with the fit in some places. Should their be different metabolites in the basis set to improve the fit? Particularly for GABA??
It looks a little better, but the aspartyl region is still quite a bit off. I suppose one thing that might help is to separate the NAA aspartyl from the NAA methyl groups (effectively, have two different basis functions), since their T2s are quite different, which would probably lead to a more pronounced amplitude mismatch between the two groups at 7T.
Stiffening the baseline up a bit (increasing the DKNTMN value in the control file) would also seem to make sense here.
Generally, GABA is not easy to reliably tease out without editing even at 7T.
Stiffening the baseline is the right thing to do in the long run, but it will necessarily result in a worse fit for the moment.
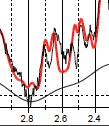
Georg is right about the T2, but the shape of things is still off (though much better than before). Especially at 2.5-ish.
Do we think that there could be an effect of the water suppression on the coupling of the 2.67 and 2.48 resonances to the 4.38 resonance? I often see issues in this region and its not just an amplitude thing.