Hi!
I really admire your efficiency in searching for papers!
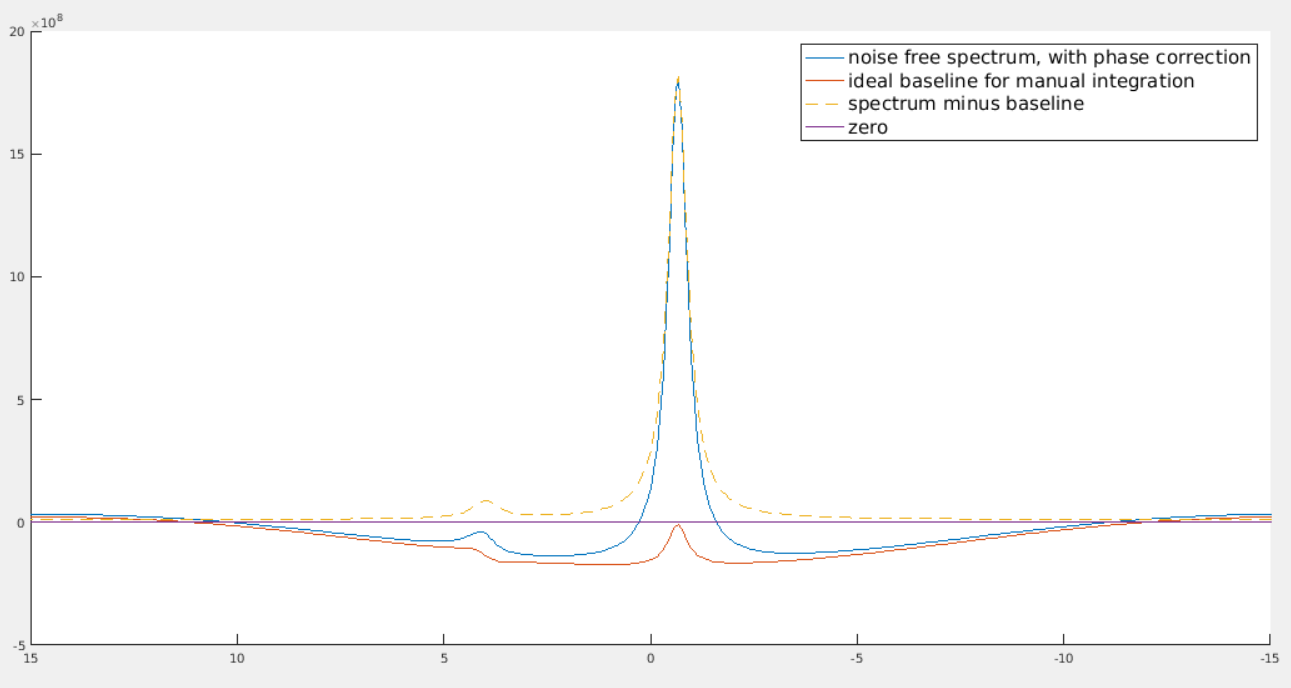
The “noise free spectrum, with phase correction” has first order phase correction. The “spectrum minus baseline” doesn’t have first order phase correction (its effect has been removed by subtracting the baseline.) The two smaller peaks are to correct the dispersion part that goes into the real part of the spectrum because of first order phase correction.
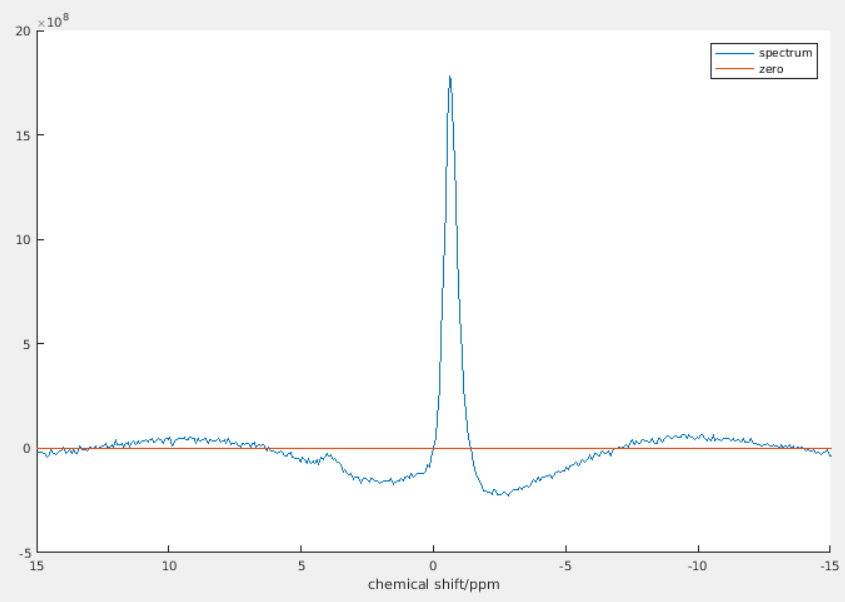
Yes, it does look much smoother if you zoom out. It becomes so unnatural because the visible ppm range is so small. As you can see, the bump of the smaller peak looks “flatter and smoother” than that of the higher peak. However, if you zoom in to the region near that smaller peak, its bump will look more obvious.
The small bump can be understood in this intuitive way: suppose the higher peak is at 172.6 ppm. After first order phase correction, the point at 172.6 ppm on the spectrum will be exactly in phase (i.e., the imaginary part is zero, the real part is equal to the magnitude). To the left of 172.6 ppm, it’s a bit off phase, and to the right, it’s a bit off phase to the other direction. Because at the point of 172.6 ppm, it’s exactly in phase, so we don’t need to correct anything for that point, i.e., baseline at 172.6 ppm should be equal to zero. Then there must be such a bump.
Yeah, I’m now looking through the spectra one by one (over 700 spectra to go through) and adjusting the polynomial for every spectrum… That being said, for some spectra with low SNR, the error is quite large (about 25%) even when two baselines look identical.
Sadly, it might be my inadequate understanding of papers. In my past experience, it happened quite a few times when I spent some time reading papers and implementing the algorithm or learning how to use their package only to find that it doesn’t work as well as expected.
By the way, does anyone share similar experiences? I don’t mean the methods in those papers are not good. Just feel that there is the risk not to get expected result after trying new (even classic, sometimes) algorithms.
And when two methods happen to work reasonably well but the results differ… Then there must be something wrong. But I don’t know what is wrong. Then I would begin to doubt “what on earth that I know is truly true?”
Anyways, I don’t have the time to try out other methods as I need to finalize the results by today. I raised this question in the beginning because I was doubting whether a polynomial can mimick an ideal baseline. It seems that the difference is just the area of that small bump. With low SNR, I guess it’s negligible.
Thank you!!