I’m dealing with MEGA-PRESS data from a Philips scan with both raw_act and raw_ref data.
I’ve followed the pipeline suggested by Gannet for the preprocessing:
For each of the step I obtained the output, but I’m not sure about the quality of my experiment.
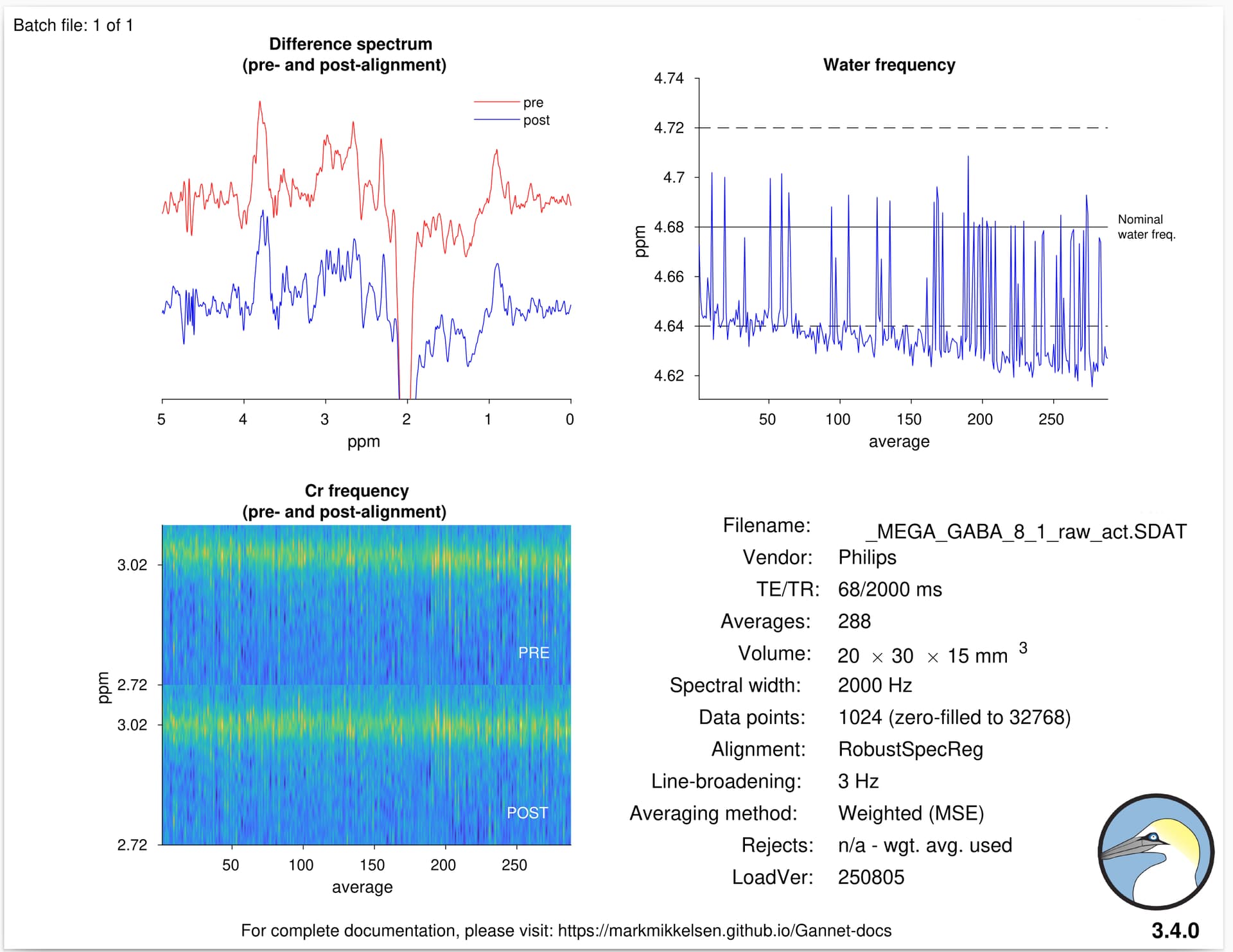
Here attached an example of GannetLoad’s output.
Could anyone give me some rule of thumb to make a sort of quality check of my data, or some general feedbacks about them?
9 ml is a very small voxel size to do any kind of MRS with, let alone spectral editing. We do not see a clearly resolved 3-ppm edited signal, so I would not recommend proceeding with this protocol.
We usually recommend starting at 27 ml and 320 transients, and then slowly working your way down from there.
I also see what looks like a systematic frequency offset (water should be at 4.68 ppm) and non-negligible frequency drift over the course of the experiments, both of which look like they’re throwing off the targeted editing pulse frequency. We’d expect the Glx signal at 2.25 ppm to be larger than it is.
We’re talking about a VOI that includes part of the left DLPFC/ACC. We usually perform SV PRESS MRS in the same VOI, this is the reason of the size.
I also noticed the drift in the water signal too, but I was unsure about the final conclusion.
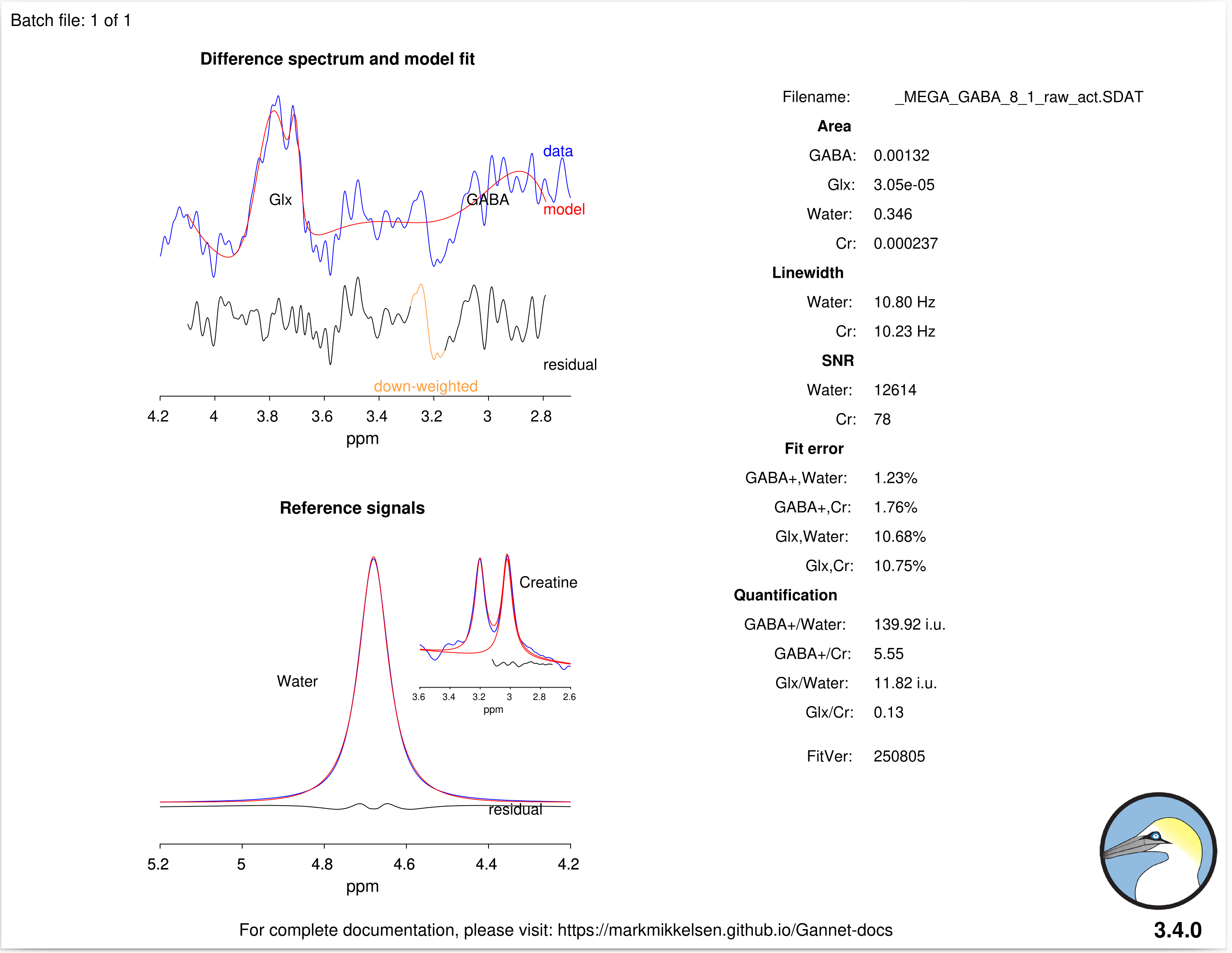
Here’s the output of GannetFit: it seems that, besides its poor quality observed in the output in GannetLoad output, the error of the fitting is relatively low (1.23% and 1.76% for GABA).
How is it possible?
No, they’re not valid at all. The Gannet fit error is only relative - it compares the standard deviation of the residual to the estimated amplitude.
But the Gannet model is not particularly smart; all it does is try and fit a Gaussian with a baseline underneath to the signal region, without any constraints. If the edited signal does not look like the expected GABA+ peak at all, the model will not be appropriate, and come up with completely unreasonable amplitude estimates.
Your estimated amplitude is gigantic (approximately 100x as big as we would expect) - look at the concentration estimates, you can’t have ~130 mM of GABA! Only because of this absurdly highly estimated amplitude, the ratio of the standard deviation of the residual to the estimated amplitude (ie the fit error) is tiny.
This is what modeling failure looks like, and I would consider neither the data nor the concentration estimates as valid.
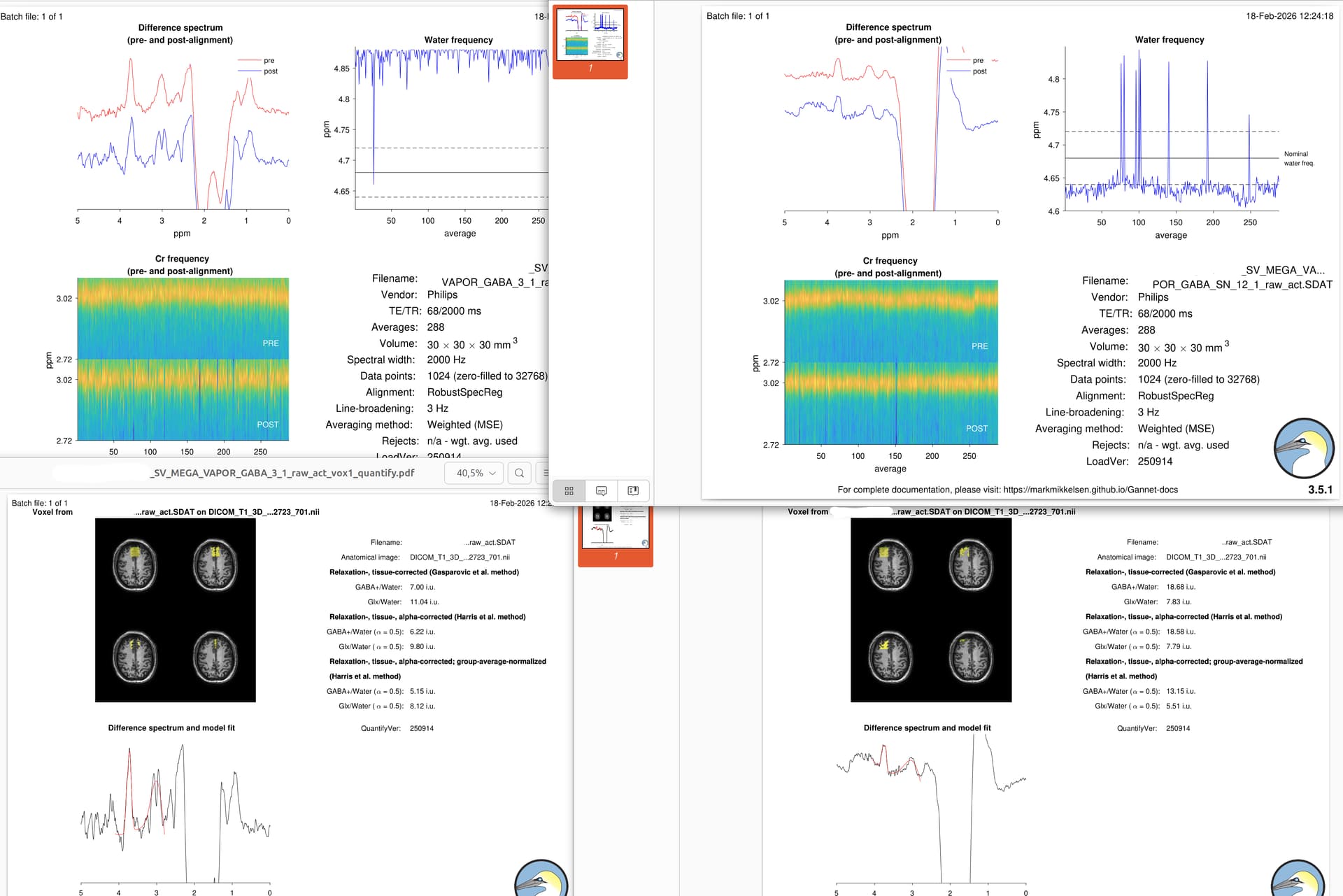
Thanks for your reply. We have now tested the sequence with a wider VOI (30x30x30) selecting the standard Philips sequence for MEGA PRESS.
We tried two different locations for the VOI: one central and another one in left hemisphere. I processed the file and I put here the Gannet’s outputs.
I think that the signal is generally better than before, but between the two spectral I would not be able to decide which one is the best (except for the drift in the water frequency in the central VOI).
Could you (or whoever) give me an advice in this sense?
I’d say this is progress (but not perfect, which may be hard to get to in this location).
These look better in terms of SNR. The one on the right has substantial lipid contamination (the large peaks between 0.5 and 2 ppm, you’ll see it even more clearly in the edit-OFF spectra) which should be avoided. You need to either move the voxel further away or change the gradient directions (more info in this thread: MEGA-PRESS Study Design Help - Study & Experiment Design - MRSHub Forum)
Linewidth is still not great (possible acceptable), but that’s not a surprise for very frontal locations.
The aligment (spectral correction) is also not optimal; there looks to be a subtraction artefact (that little dip in the 3-ppm signal in the left spectrum should not be there). You can try the other alignment options that Gannet offers.
I agree that positioning the VOI at the frontal level is challenging. This is why we attempted to place it in a more central position (the spectra on the left) instead of the left position (shown on the right in the previous image).
Since you commented on the more central VOI, do you think it is the better option of the two? Also, would choosing the central one still be the best solution despite the very strong water drift signal?
In any case, I’ll try with another alignment, as suggested.
I don’t think you have an issue with drift rather than a systematical offset.
It is a little concerning that the water frequency is displayed at >4.85 ppm, I agree - can you share screenshots of the PDF protocol printout? There’s a setting in there to confirm/update F0 prior to starting the scan but I can’t recall the name