Firstly, thank you so much for providing this user-friendly software! It is very comprehensive and easy to follow. I went through the other posts and found nothing matching my questions. I would appreciate it if you could help me to solve my problem.
Part one:
I have read the “Contribution of macromolecules in Experts’ consensus recommendations” article, but I do not understand clearly what does this sentence mean?
" If the objective of a study is to determine metabolite concentrations and ratios, then the MM contribution has to be removed or included in the basis set used for quantification, especially for TEs below 80 ms. "
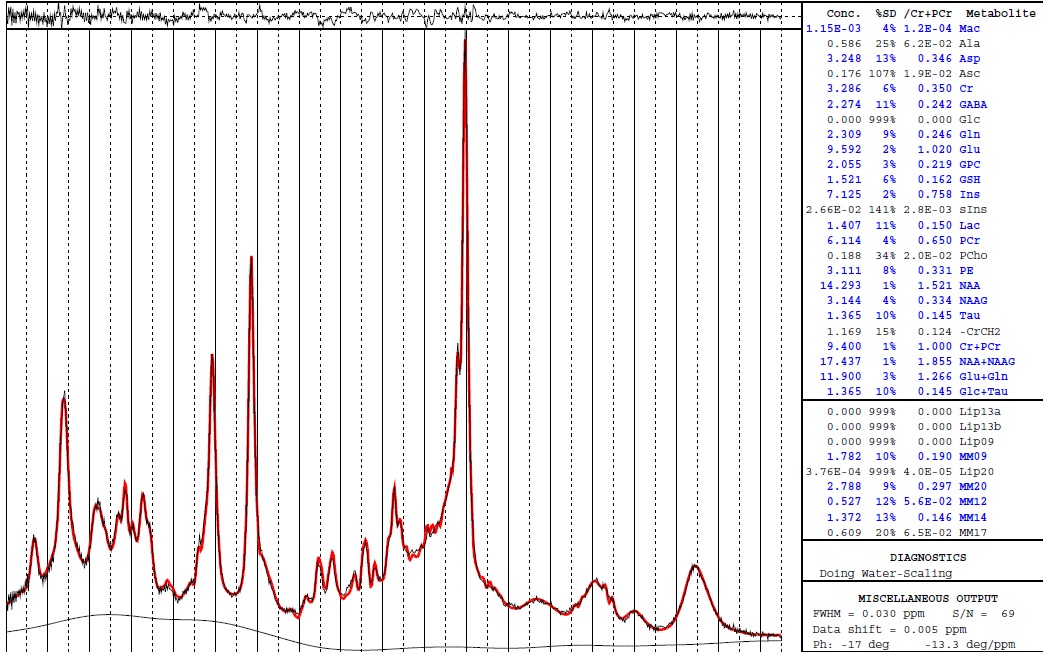
Does this sentence mean after I analyse the spectra with LCmodel, I should not see the concentration of lipids and MM in the box below the metabolite concentration box? What if I use the basis set in which the MM measurement is included? Should I not still see the MM quantification?
If yes, I think I need to put NSIMUL=0 in the lcmodel control file to remove MM quantification. Yes?
Part two:
If I need to delete MM concentration to determine metabolite concentration accurately, I do not know how I can do it with Osprey. (1) I ticked off and on the 'add MM and Lip basis functions to fit" option in creating Job,(2) I unticked MMs in the selected Metabolites, (3) I changed the NSIMUL=0 in the control file that was generated by Osprey automatically, but The MM concentration was still there after I did all these steps.
To clarify the sentence you quoted, it’s not really that the MM signal needs to be removed altogether, more that the MM contribution to metabolite resonances must be removed. Or, to state things another way, you must attempt to account for the MM signal that will be present in your data so that the metabolite estimates are more accurate. One way to go about this is to include some MM model functions in your basis set, which appears to be what’s happening in the attached image, and in that case, a non-zero MM amplitude just means the MM resonance has been accounted for, which is a good thing.
Hopefully this also answers part 2? Perhaps someone more familiar with LCModel could say more about the optimization of this setup, but the fit looks good to me!
just an important detail, when we wrote this paragraph it was to highlight the fact that MM need to be included in the basis set even at longer echo time. If your TE is not longer than 80ms then you should also include MM in your basis set.
best
cristina
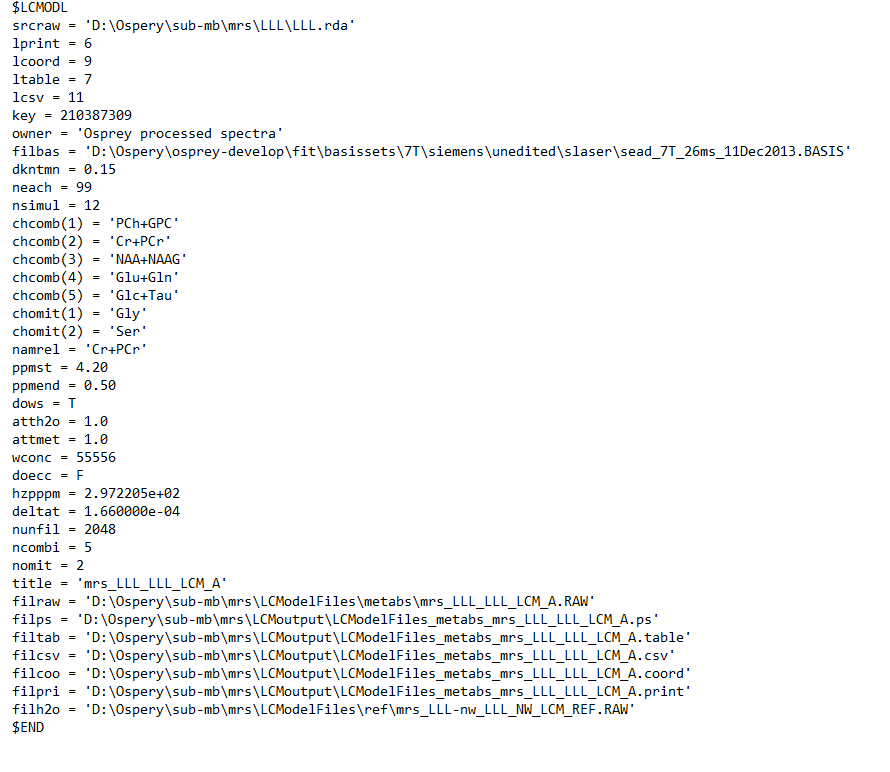
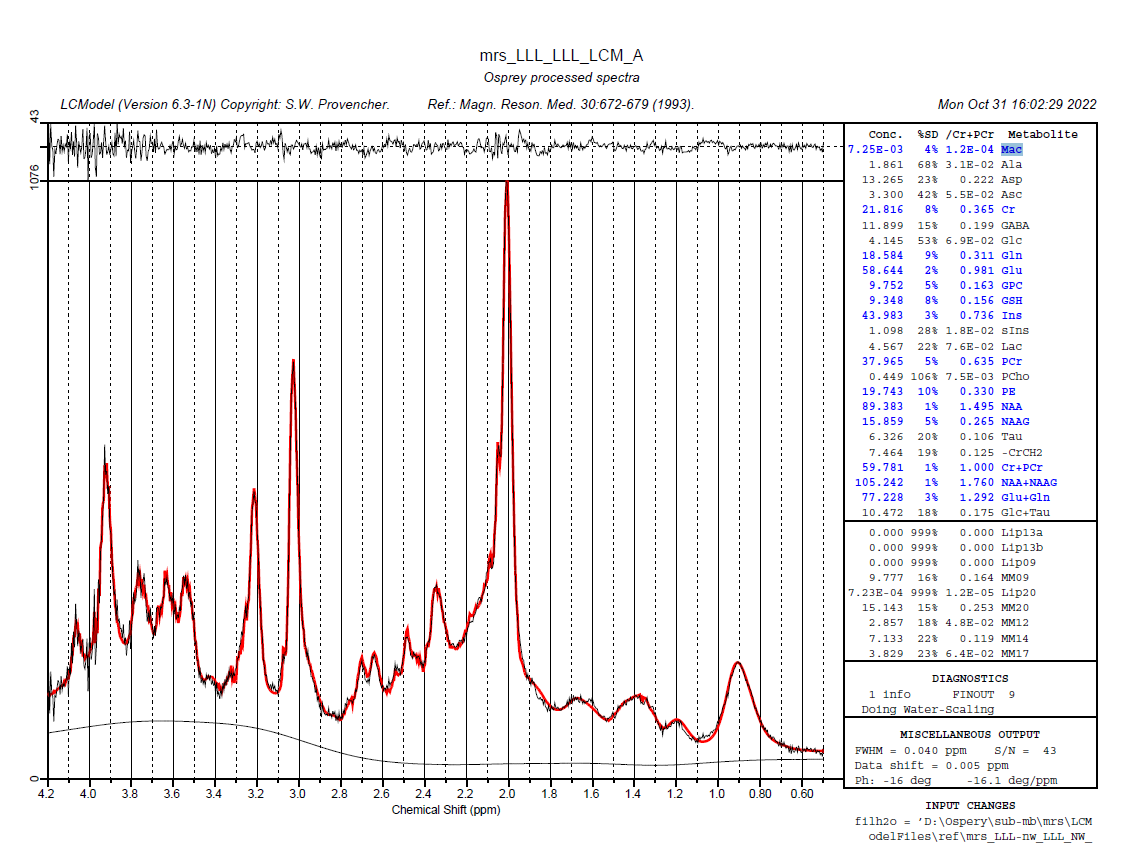
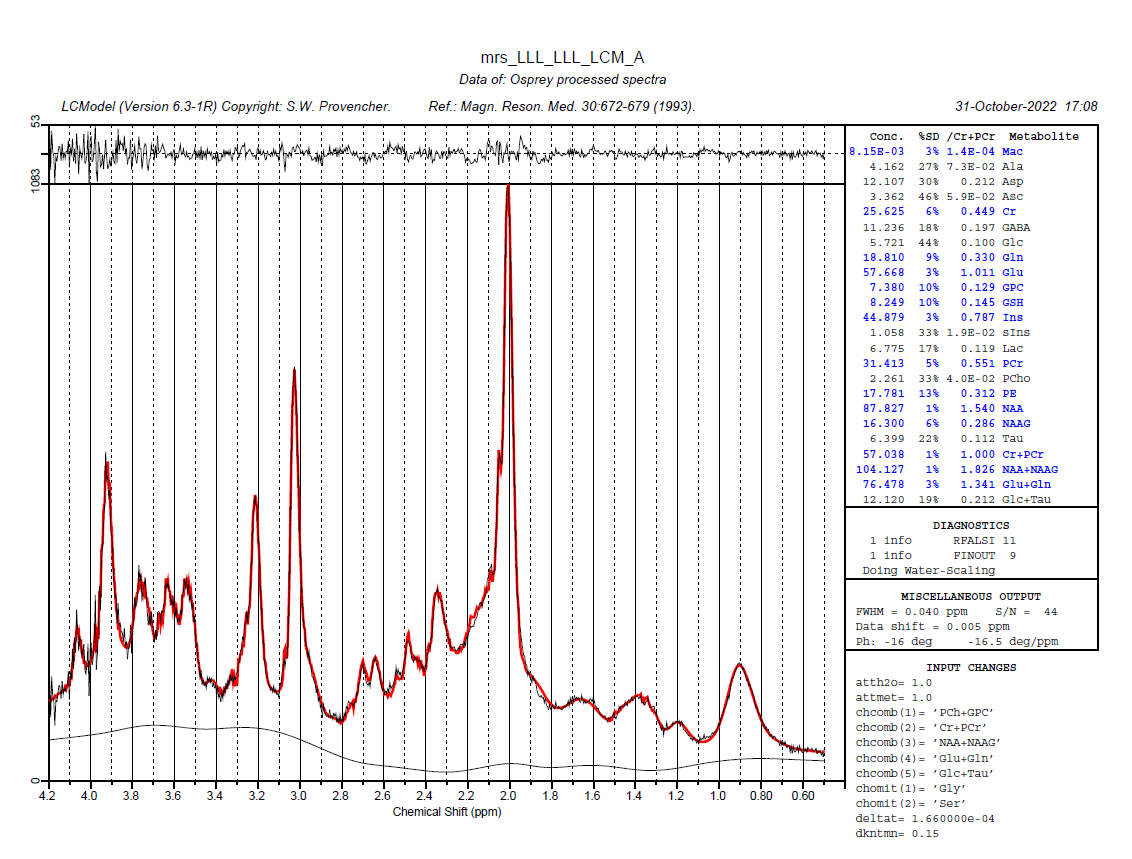
Can you post the exact control file that Osprey generates? Are you pointing to a template control file in the job file? I’m seeing that the fit includes both a measured macromolecule background (Mac) and individual components (MM09-17). That’s a few too many degrees of freedom for my taste - @cudalbu my understanding is that EITHER simulated OR measured MM is acceptable, but not both together? We might need to tweak the control file a little.
NSIMUL=0 should indeed at least prevent LCModel from including the MM09-17 components, but for some reason that hasn’t worked here, so there might be other settings in the control file that override it.
The TE I used was 26 ms, I asked the basis set developer and he said the measured macromolecule is included in the basis set. Sorry for my foolish question in advanced. When we see the concentration of Mac and MM09,MM20,MM12,MM14,MM17 in the LCmodel analysis file, it means the measurement of MM was included in the basis set? I mean how can I understdand that my analysis and basis set that i am used in terms of metabolites concentration and MM estimation are accurate? Should I see the concentration of Mac and MM09,MM20,MM12,MM14,MM17 in LCmodel or not?
just pay attention that when you have Mac estimated in the table it means that a Mac signal is already included in the basis set. Then when you have also the Lip and MM below Glc+TAu it means that in tot of the Mac in the basis set you also fit hem a second time using the simulated MM+Lip from LCModel - so you fit twice the same. it is advised to use only one, better the one measured
best
cristina
Yes he is right the MM are included in the basis set since you can see the “signal/metabolite” called Mac in the table in the right part of the screen - you can also plot in the pdf all your metabolites from your basis set using the command NEACH =999 @simicic can you please confirm? i’m not sure about the command anymore
for teh other question i just answered below - Mac and MM09,MM20,MM12,MM14,MM17 are two ways to fit the same thing
Mac - when you have the signal of macromolecule sin your basis set - it can be measured in vivo or simulated but it is an entire signal composed of all macromolecules resonances
MM09,MM20,MM12,MM14,MM17 and Lip - are individual resonances that are simulated by LCModel to fit the same thing - the macromolecules however they are not complete (the number after each letter represents the ppm region where they are located, and you can see that they do not cover the entire ppm range where the macromolecules are supposed to be)
you should only use one of them - the Mac or the LCmodel MM and Lip
best
cristina
Hi Georg @admin,
Can you comment further on what you mean about the degrees of freedom in this context? What happens if both measured and simulated MM are included?
I agree with both Cristina and Georg, you either go with simulated MM (the components estimated by LCmodel) or with measured MM included in your basis-set (in your case that is Mac that you see in the LCModel quantification fitting), not both. If you have the measured MM its always better to use them instead of the LCModel components. So in your case I think the NSIMUL=0 in the control file and the basis set you are already using is a good approach. Moreover, as Cristina mentioned if you include NEACH=999 to your control file, in the LCModel pdf you will additionally be able to see the individual fit of every component of your basis set (both individual metabolites and MM). Therefore if you look at your quantification without NSIMUL = 0, you will see that you are fitting your Mac (basis set MM) and the LCModel MM components to the same resonances, which indeed may lead unnecessary overfitting.