Hi everyone,
I’m encountering an issue with FSL-MRS during the quantification step, and I’d appreciate any advice on resolving it. The error message indicates an unusual echo time and a missing water scaling metabolite. Here’s what I’ve done so far:
-
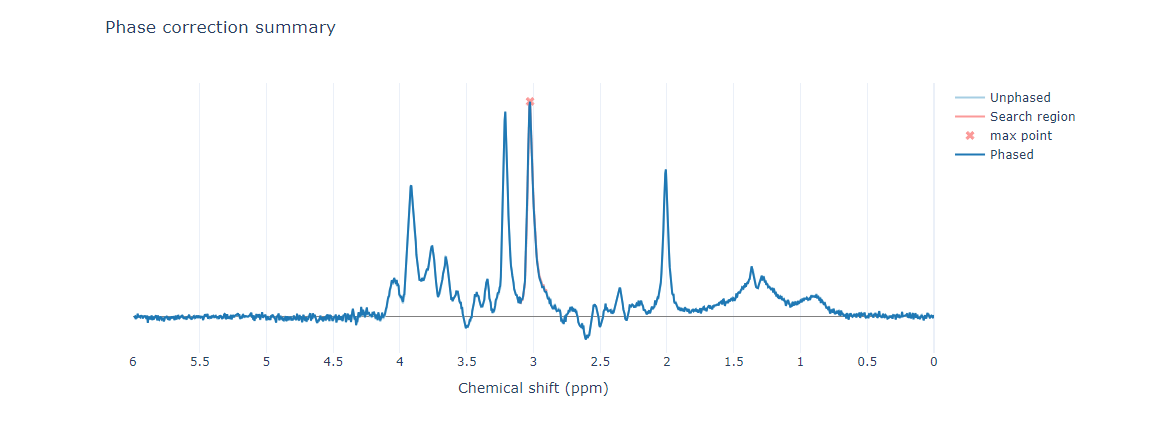
Preprocessing: I used fsl_mrs_preproc to preprocess my data, which seemed to work well, although the NAA peak appears slightly lower than expected.
-
Basis Set Simulation: I simulated my basis set with the command:
fsl_mrs_sim -m Glu,H2O -o basis_output -e 30
I generated a JSON file based on my MRS parameters (MRS_parameters.pdf (631.6 KB), but I’m not entirely sure if it’s accurate eja_svs_mpress_on_to_simulate_basis_set.txt (1.2 KB) -
Basis Set Output: This generated separate basis sets for H2O and Glu in .txt, .json , and .raw formats, as separate files for water and Glu.
For fitting, I used:
fsl_mrs --data “path to my metab.nii file”
–basis “path to my Glu.txt”
–output “”
–h2o “path to my wref.nii”
–report
- Error Received:
NoWaterScalingMetabolite: No suitable metabolite has been identified for water scaling.
QuantificationException: No metabolites in FSL’s dictionary of water scaling metabolites are in the basis set. Please specify a water scaling metabolite manually (–wref_metabolite). Also specify --ref_protons and --ref_int_limits.
I’ve tried adjusting the command without success, and I also attempted to combine the Glu and H2O basis sets manually, but this hasn’t worked either. Does anyone have insights on correctly specifying the water scaling metabolite or suggestions for troubleshooting?
Thank you!
P.S. I forgot to add some important information: the sequence is MEGA-PRESS with water suppression. The data file structure includes 4 On sequences with 64 pulses each and 1 Off sequence. Currently, for my analysis, I am using only one On and one Off sequence because I’m uncertain whether I should average the raw data, the preprocessed sequences, or the final fitting outputs.
Additionally, it seems that the peak is being reduced during alignment.