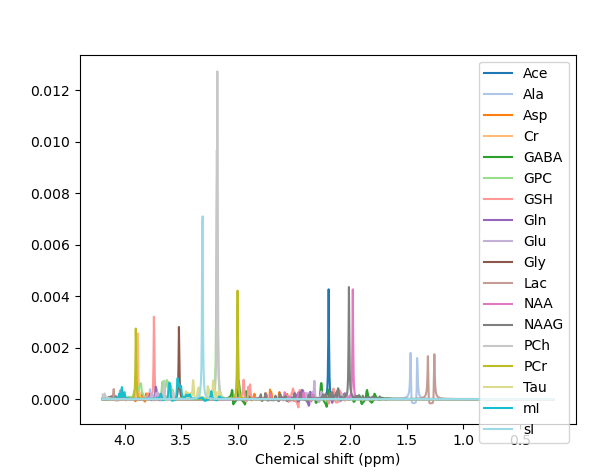
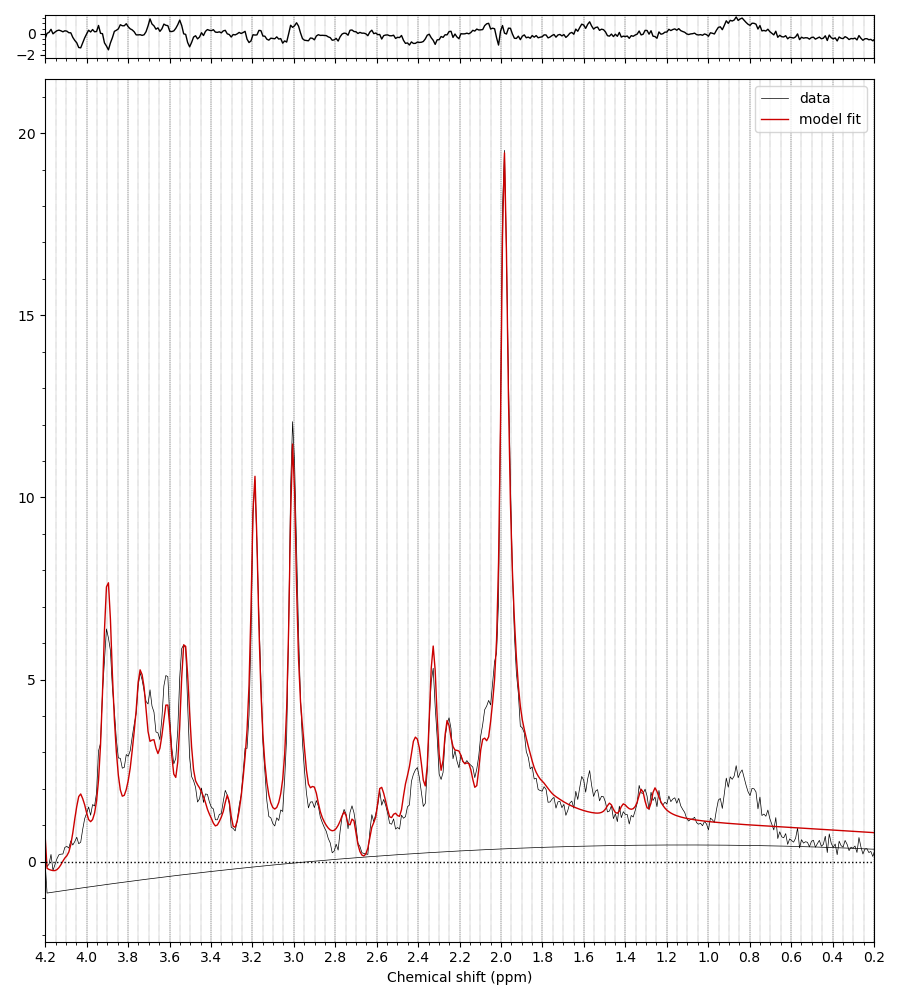
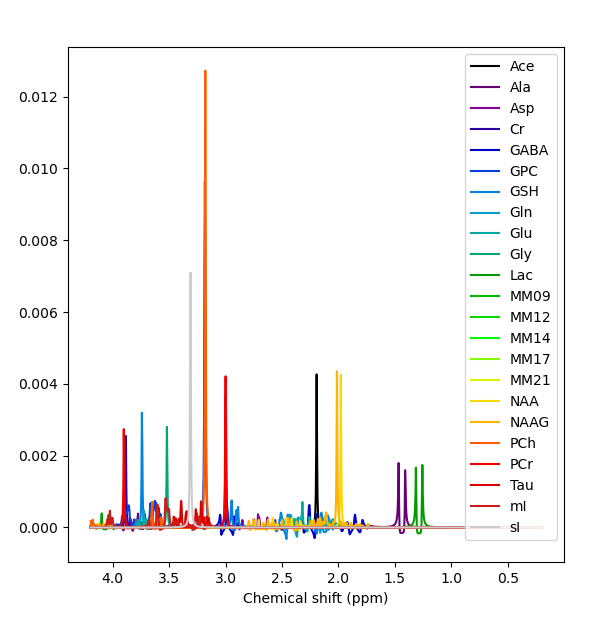
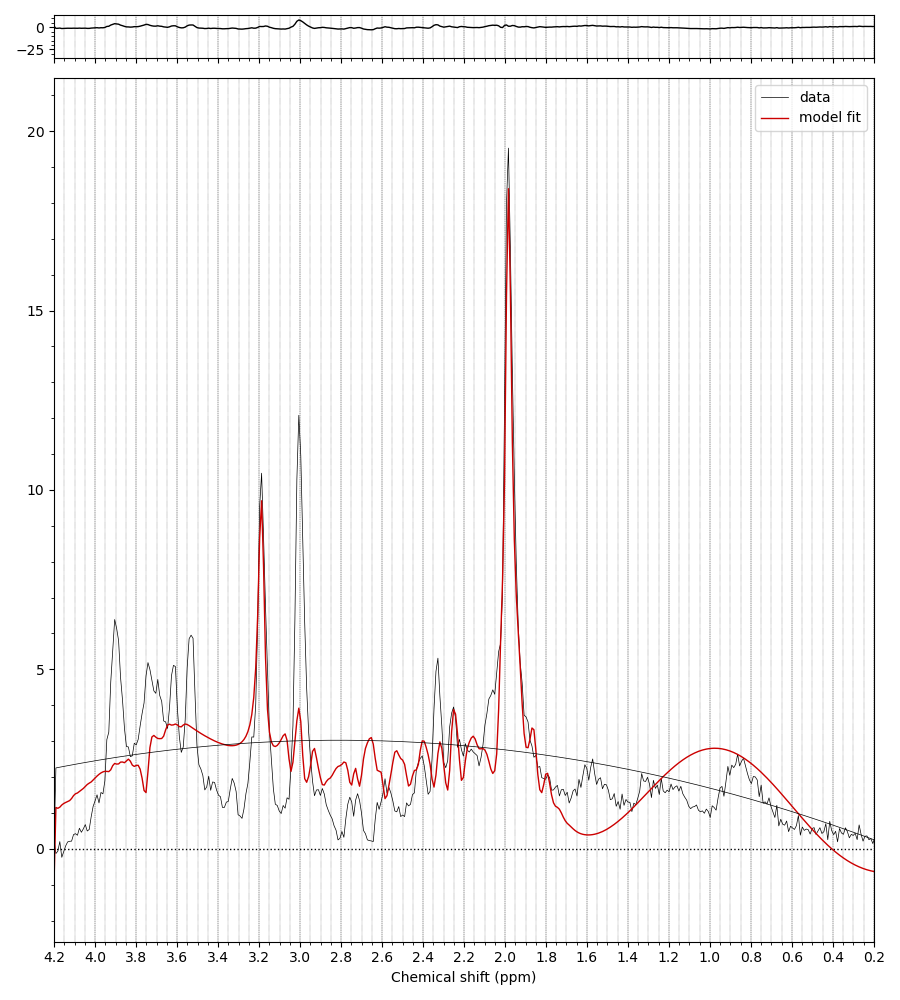
We’ve got some online resources that can help with the understanding of what’s in a basis set, and how to create one. See these videos and the last bit of the online practical.
Traceback (most recent call last):
File "/usr/local/fsl/fslpython/envs/fslpython/lib/python3.8/site-packages/fsl_mrs/core/basis.py", line 302, in _resampled_basis
basis = misc.ts_to_ts(self._raw_fids,
File "/usr/local/fsl/fslpython/envs/fslpython/lib/python3.8/site-packages/fsl_mrs/utils/misc.py", line 210, in ts_to_ts
raise InsufficentTimeCoverageError('Input data covers less time than is requested by interpolation.'
fsl_mrs.utils.misc.InsufficentTimeCoverageError: Input data covers less time than is requested by interpolation. Change interpolation points or dwell time.
During handling of the above exception, another exception occurred:
Traceback (most recent call last):
File "/usr/local/fsl/fslpython/envs/fslpython/bin/fsl_mrs", line 426, in <module>
main()
File "/usr/local/fsl/fslpython/envs/fslpython/bin/fsl_mrs", line 229, in main
conjugated = mrs.check_Basis(repair=True)
File "/usr/local/fsl/fslpython/envs/fslpython/lib/python3.8/site-packages/fsl_mrs/core/mrs.py", line 588, in check_Basis
basis = self.basis
File "/usr/local/fsl/fslpython/envs/fslpython/lib/python3.8/site-packages/fsl_mrs/core/mrs.py", line 231, in basis
return self._basis.get_formatted_basis(
File "/usr/local/fsl/fslpython/envs/fslpython/lib/python3.8/site-packages/fsl_mrs/core/basis.py", line 224, in get_formatted_basis
formatted_basis = self._resampled_basis(1 / bandwidth, points)
File "/usr/local/fsl/fslpython/envs/fslpython/lib/python3.8/site-packages/fsl_mrs/core/basis.py", line 307, in _resampled_basis
raise BasisHasInsufficentCoverage('The basis spectra covers too little time. '
fsl_mrs.core.basis.BasisHasInsufficentCoverage: The basis spectra covers too little time. Please reduce the dwelltime, number of points or pad this basis.
In that thread, the person had a similar error, but it was fixed by using the same bandwidth as the one they downloaded. I am doing that… so I’m not sure what I’ve done wrong.
What bandwidth (dwelltime) and how many points does your data have? If the product of dwelltime * points of your data is greater than dwelltime * points of the basis then you will get this error.
What ends up weird? Is it that the MM are at the wrong frequency? If so try using basis_tools conj before adding the MM. Or is it that the MM are very narrow? If so you can specify Lorentzian and Gaussian broadening in the call to add default MM
% basis_tools add_set --help
usage: basis_tools add_set [-h] (--add_MM | --add_MM_MEGA | --add_water) [--gamma GAMMA] [--sigma SIGMA] file output
positional arguments:
file Basis file
output Output location, can overwrite.
optional arguments:
-h, --help show this help message and exit
--add_MM include default macromolecule peaks
--add_MM_MEGA include default MEGA-PRESS macromolecule peaks. This option is experimental!
--add_water include water peak.
--gamma GAMMA Peak widths (lorentzian) in Hz.
--sigma SIGMA Peak widths (gaussian) in Hz.
What does the final basis look like? One issue is that you might need to run basis_tools conj again. Sorting out the basis set conventions is on the to-do list, pending a new standard that various people ware working on.
There appears to be a couple of things going on here with the basis.
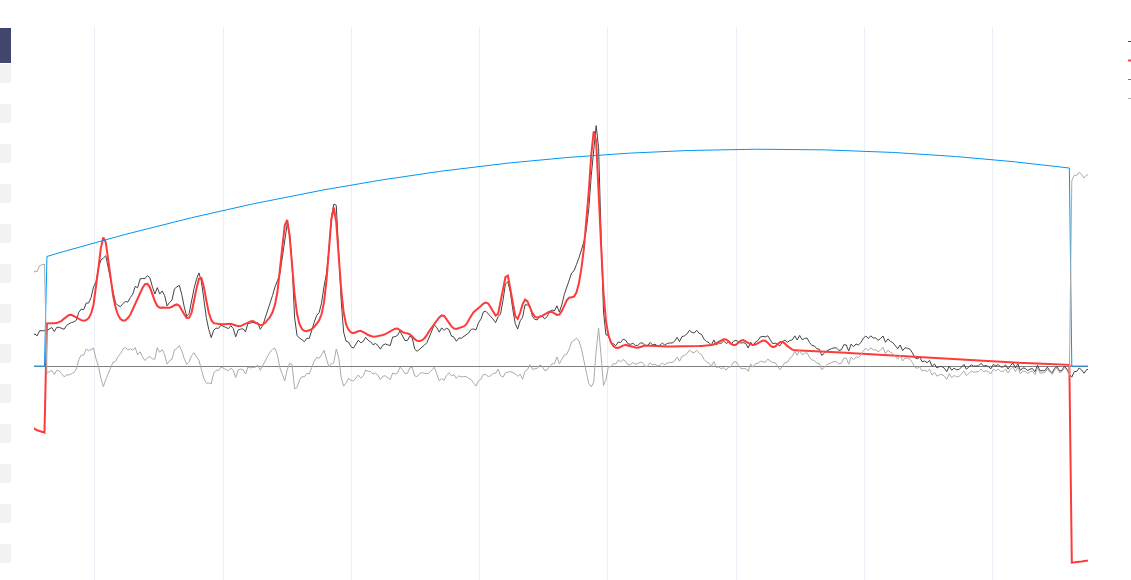
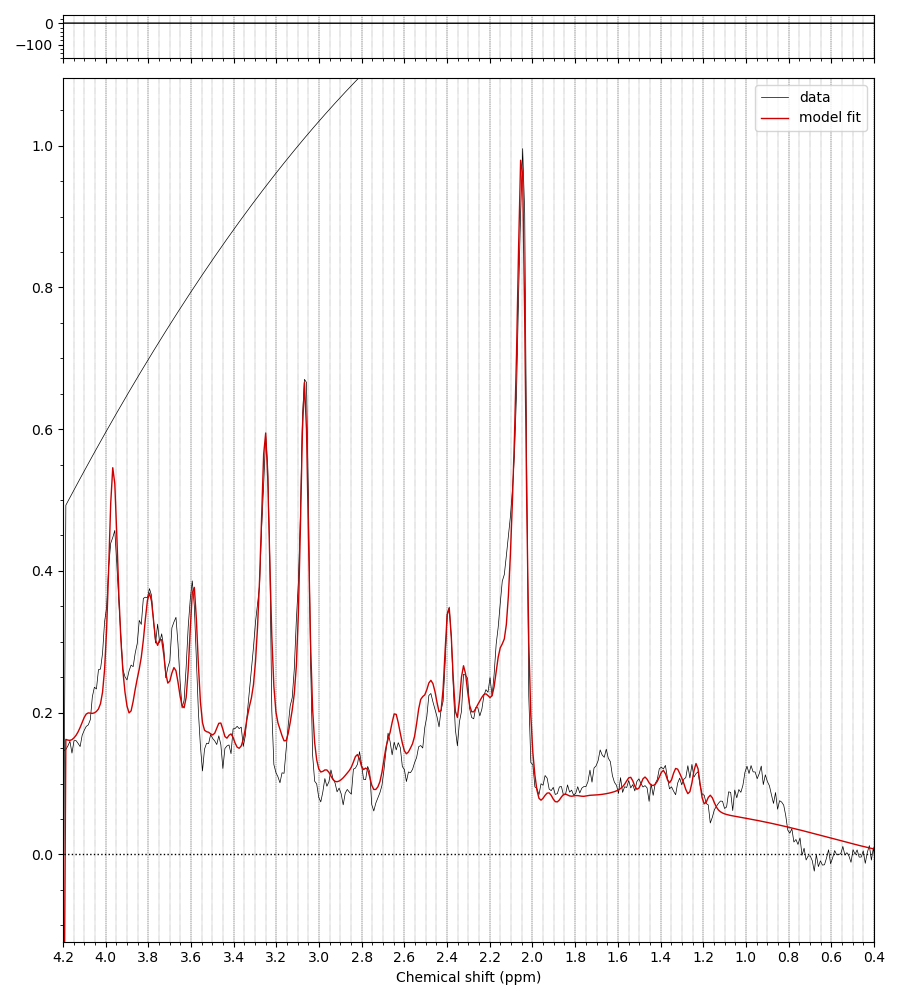
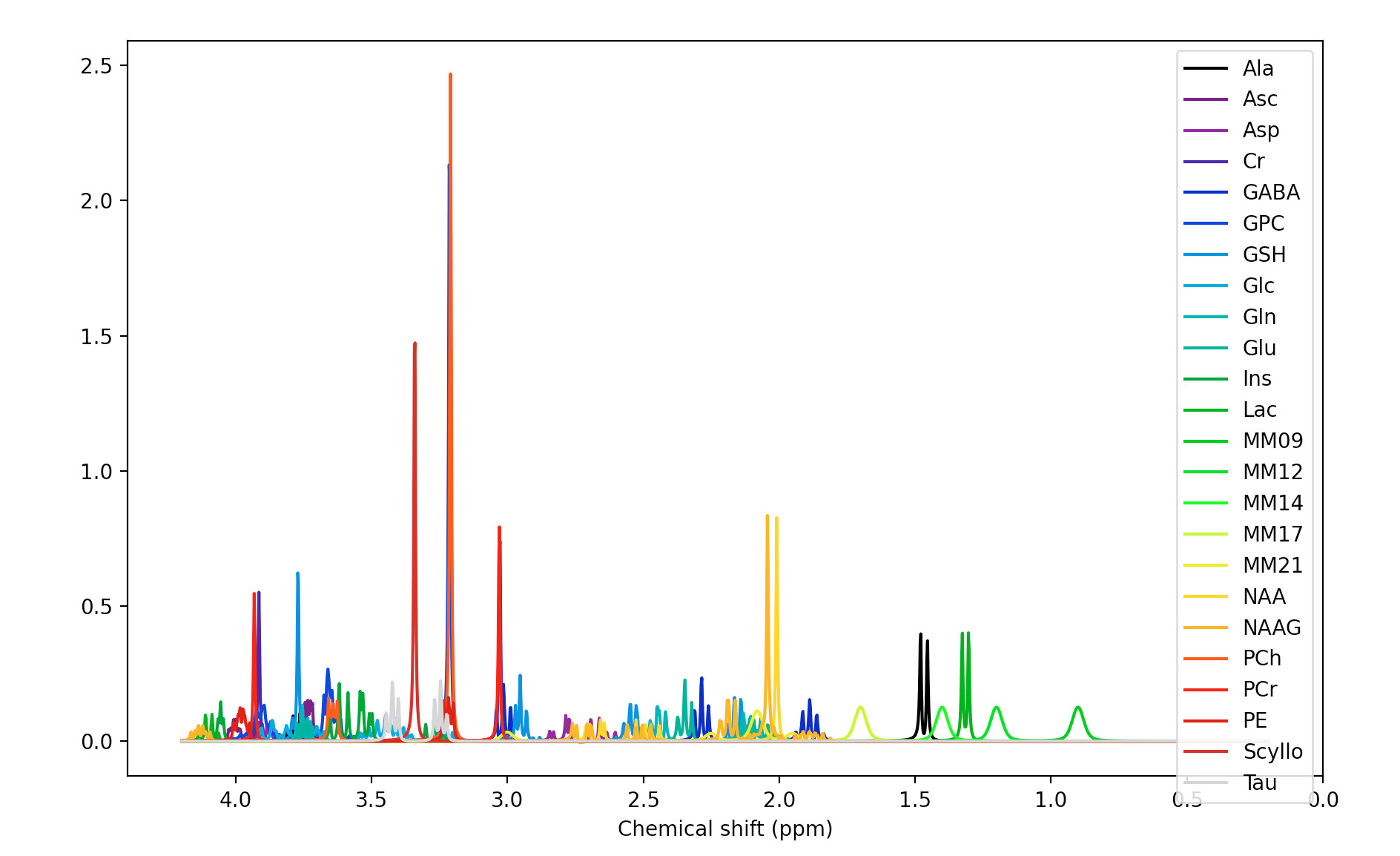
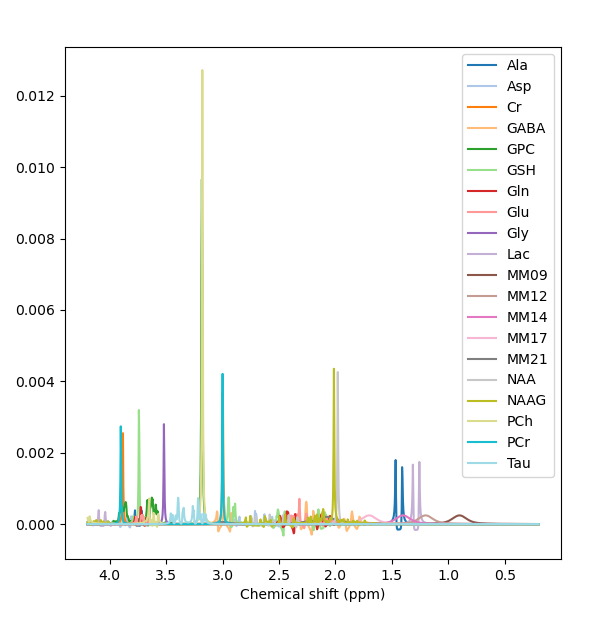
The MM appear to have been applied when the basis set is conjugated. Precisely what commands did you run to get the first figure in the previous post?
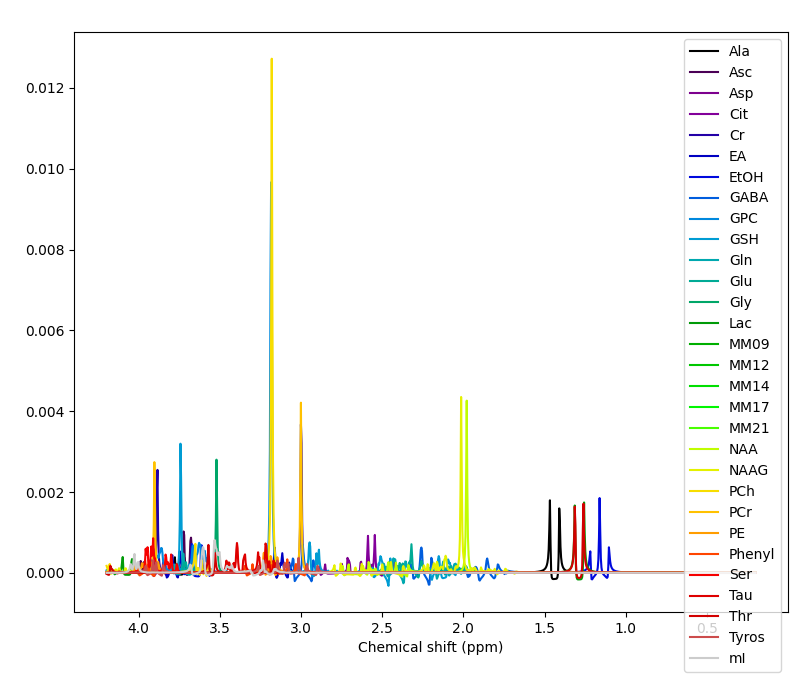
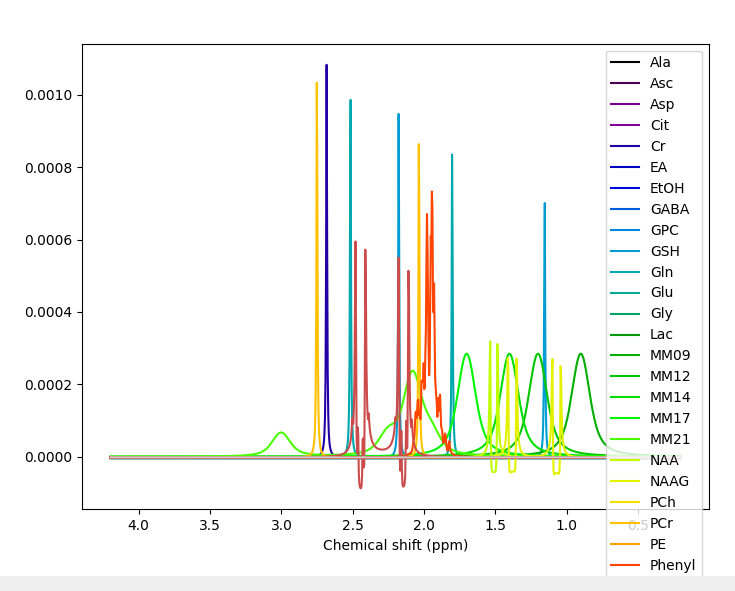
That basis set comes with many metabolites, I would suggest trimming it down. To precisely what remains a debated question, but perhaps remove Tyros, Thr, Ser, Phenyl, PE, EtOH, EA, Cit and Asc to start.
Those commands look right, but you are on quite an old version of FSL-MRS now. You have the version that comes with the ‘current’ (> year old) version of FSL. However, as a newer tool in the FSL stable development has kept going. Could you update using the instructions here? Installation Instructions — FSL-MRS 2.0.7 documentation
Re the spec2nii. You are not the only one. It’s relatively near the top of my things to do list. I think @mmikkel has a solution.
Thanks for bringing this to my attention. It seems this is the version that is installed with the most recent version of FSL? (I even re-installed to see if it updated, but it stays the same)
The best solution for me was to create a conda environment and then install using conda.
The reason I have to create a conda environment is that, if I install with the base environment, it still defaults to /usr/local/fsl/bin/fsl_mrs instead of the conda updated version
Ok, everything is up to date (removed all traces of what FSL default installs w.r.t. fsl_mrs and created an virt environment and installed in that using conda)
fsl_mrs -v gives 2.0.8
spec2nii -v gives 0.4.9
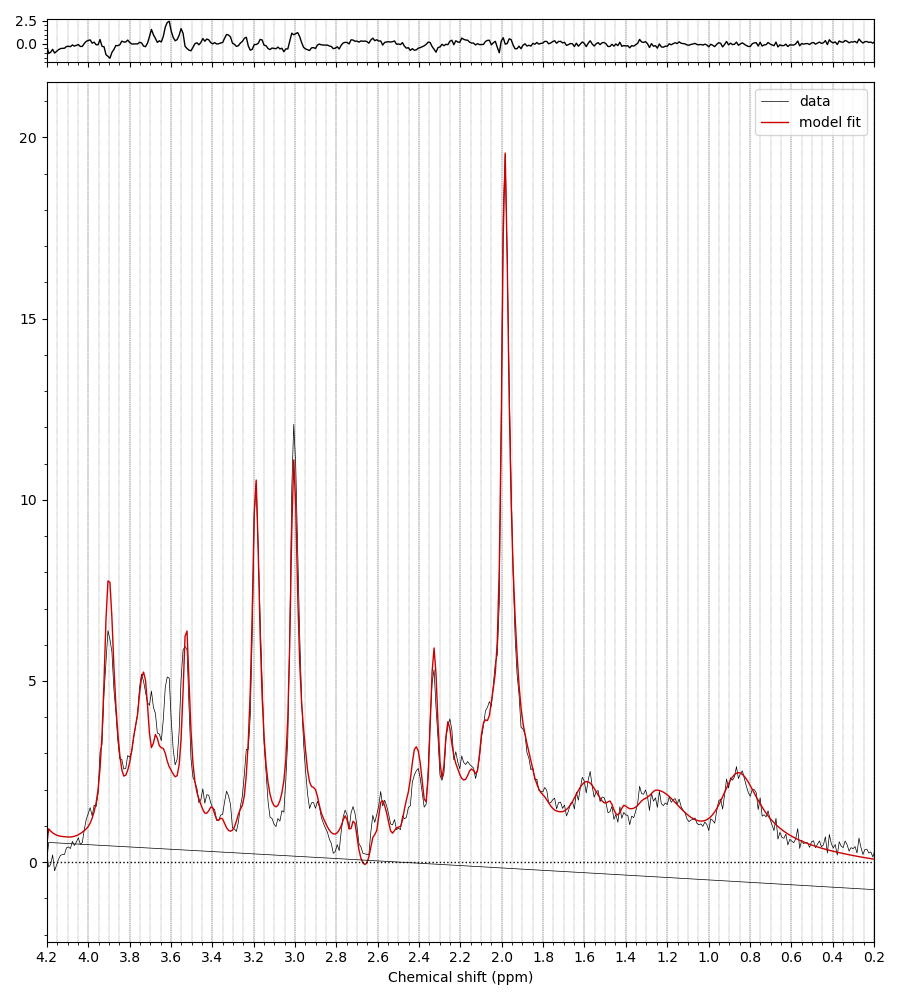
I have processed my data according to the tutorial (FSL-MRS Practical)
Yes, right the MM have been added with the wrong handedness convention. This is unfortunately a bit of a mess currently in FSL. All this is hopefully going to get sorted soon when @Helge establishes a new standard for basis sets :).
For now take the first basis set you have just shown me (without MM), run the conjugation step, then the add MM, then the conjugation step again on the basis with MM added. You should then see the (broad MM) peaks in the right positions roughly 1-2 ppm. Something like this: