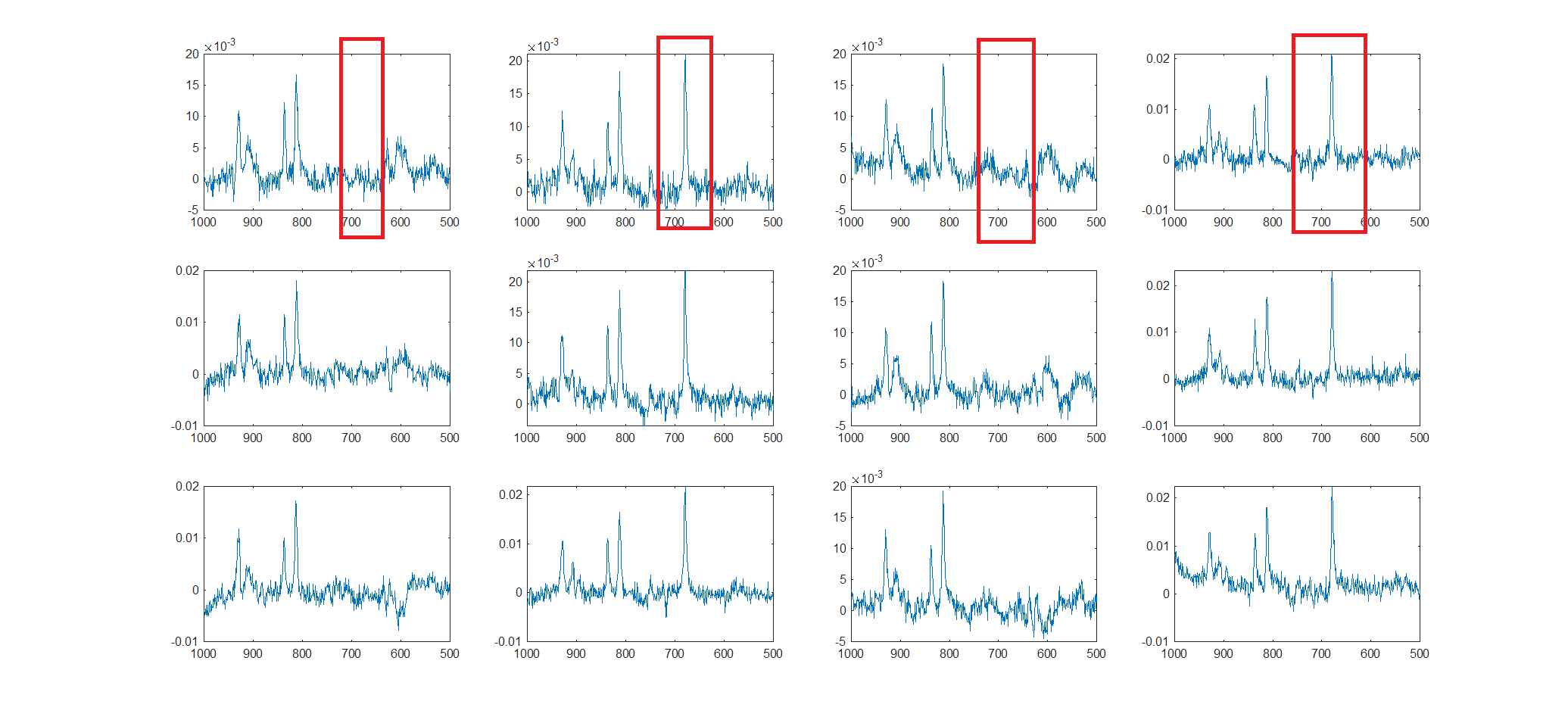
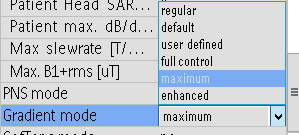
Hello everyone! I’m immensely grateful to this community for the generously shared knowledge! I apologize in advance if the following questions are basic!
I am new to the acquisition of PRESS (Glx) & MEGA-PRESS (GABA+). I plan to use Osprey to analyse and report PRESS and MEGA-PRESS data acquired from the same anatomical areas based on the recommendation given here.
I am acquiring spectroscopy data from cooperative clinical patients with severe symptoms in a hospital setting. We are allowed limited time in the scanner (total scan duration not to exceed 55 minutes), including several compulsory protocols (21 minutes, not to be modified).
Within the limited time left (34 minutes approx.), based on our clinical intervention, I wish to acquire single-voxel PRESS and MEGA-PRESS data from two anatomical sites: left dorsolateral prefrontal cortex (dlPFC) and left temporoparietal junction (TPJ). We are using Philips 3T Ingenia CX system v5.8 with a 32-ch head coil (I didn’t find an option to export protocol as a pdf!).
Based on the extensive feedback in this thread and this reference, we have come up with the following sequences. Given the time crunch, I need advice on optimizing acquisition parameters.
-
PRESS SEQUENCE FOR
-
Left dlPFC
-
voxel size: 20x15x20 mm=>6ml
-
Total Averages=64 [16 (No. of Signal Averages) x 4 (Dynamics)]
-
TR=3000 ms / TE=68 ms
-
-
Left TPJ
-
voxel size: 20x20x20 mm=>8ml,
-
Total Averages=64 [16 (No. of Signal Averages) x 4 (Dynamics)]
-
TR=3000 ms / TE=68 ms
-
-
-
MEGA-PRESS SEQUENCE
-
Left dlPFC :
-
voxel size: 32x27x32 mm=>27.64ml
-
Total Averages=144 [16 (No. of Signal Averages) x 9 (Dynamics)]
-
TR=2000 ms / TE=68 ms
-
-
Left TPJ : voxel size:
-
30x30x30 mm=>27ml
-
Total Averages=144 [16 (No. of Signal Averages) x 9 (Dynamics)]
-
TR=2000 ms, TE=68 ms
-
-
-
Short TE scan for
-
Left dlPFC
-
Voxel size: 32x27x32 mm=>27.64ml
-
Total Averages=32
-
TR=2000 ms / TE=30 ms
-
-
Left TPJ
-
Voxel size: 30x30x30 mm=>27ml
-
Total Averages=32
-
TR=2000 ms / TE=30 ms
-
-
-
Water Un-Suppressed scan for
-
Left dlPFC
-
voxel size: 32x27x32 mm=>27.64ml
-
Total Averages=32
-
TR=2000 ms / TE=68 ms
-
-
Left TPJ
-
voxel size: 30x30x30 mm=>27ml
-
Total Averages=32
-
TR=2000 ms / TE=68 ms
-
-
I have attached the concatenated exam cards for all these scan sequences as a single document. The compulsory protocol includes T1 weighted imaging (TR=6.5ms / TE=2.9 ms; exam card included as attachment). I will be using this for in-voxel tissue segmentation.
Anushree_Bose_EXAM_CARDs.txt (27.9 KB)
We prefer to acquire PRESS over MEGA-PRESS.
As I have limited time to dedicate to spectroscopy (34 minutes) in a 55-minute scan, any advice on what to keep and chop is truly appreciated! A few questions:
-
What matters more for MEGA PRESS: acquiring more signal averages or accommodating a water un-suppressed scan and short TE scan? Is there a trade-off? Recommended number of averages is 300+, but we are acquiring 144! due to time constraints as we are accommodating Shoet TE scan and water unsuppressed scan.
-
Should TR be kept same across PRESS, MEGA PRESS and Short TE? Is it okay to use the same Short-TE scan (TR=2000ms/TE=30ms) for both PRESS (TR=3000 ms/TE=68 ms) and MEGA PRESS (TR=2000 ms /TE=68 ms) if targeting the same anatomical areas?
-
Is it correct or incorrect to vary the voxel sizes between PRESS, MEGA PRESS and Short-TE if targeting the same anatomical region? At present, I have kept the voxel sizes as same for Short-TE and MEGA PRESS but smaller for the PRESS sequence.
-
Are start-up scans (instances where complete pulse sequences is run without acquiring data in order to establish steady state) or dummy scans mandatory? If yes, are 4 ‘start-up scans’ enough?
If the time limit is too constraining to acquire sufficient averages along with Short-TE / Water Unsuppressed scans, I am open to dropping either left dlPFC or left TPJ. However, given the clinical context and relevance of the study, retaining both sites is desirable.
Looking forward to improving our efforts with your input!
Thanks and regards,
Anushree