Dear All,
Hi,
I get water FWHM a negative number while analyzing multi voxel MRS (GE pfile)
some thing like:-0.007827143 what can be the broblem??
Regards,
Neda
Dear All,
Hi,
I get water FWHM a negative number while analyzing multi voxel MRS (GE pfile)
some thing like:-0.007827143 what can be the broblem??
Regards,
Neda
Hi @NNeda,
Any chance you could share some output (eg, from tarquin’s output_pdf option), so we can better grasp what might be the problem?
It could be that your NAA peak is inverted (perhaps due to inverted residual water, or some other phasing issue), but without seeing a spectrum this is just speculation…
Alex.
Dear Alex,
Thank your very much for your reply. I have attached the PDF and excel file
pdf1.pdf (37.8 KB)
csv.csv (1.7 KB)
and the other question I have is about creatine…is the creatine icluded in TARQUIN basis set, itself the total creatine ??
BEST REGARDS,
NEDA
Dear Neda,
Thanks. It looks like Tarquin isn’t finding (or using) your water-unsuppressed reference data. Do you know if this is being acquired by your sequence? For standard single voxel MRS on GE this happens automatically, but this is not necessarily the case for other sequences on that platform…
Your basis set also looks suspiciously sparse, but that’s a separate issue; how are you invoking Tarquin? Through the gui, or command line? Most of Tarquin’s built-in basis combinations are fairly comprehensive, so it’s probably best to start with one of those (eg, 1h_brain). In that case you could get separate values for Cr and PCr, and could take the sum of these to be “total creatine”. Your current basis set only explicitly models the first (in practice, it’ll probably capture a bit of both).
Alex.
Dear Alex,
My Acquisition is on GE scanner(discovery 750w 3T) and as you said yes the unsuppressed water is automatically acquired but at the beginning of MRSI the scanner also it gives
the water FWHM so it means that it’s acquired for MRSI too,
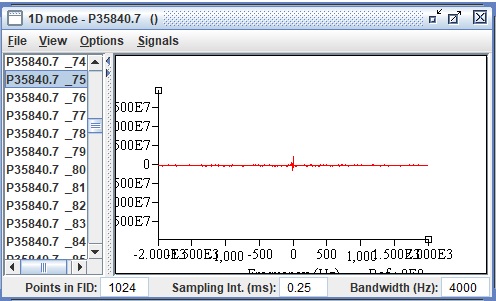
I think.Besides, I uploaded the pfile in jmrui and it separated the unsuppressed water and I concluded that it’s aquired. But on JMRUI the signal is so weak.
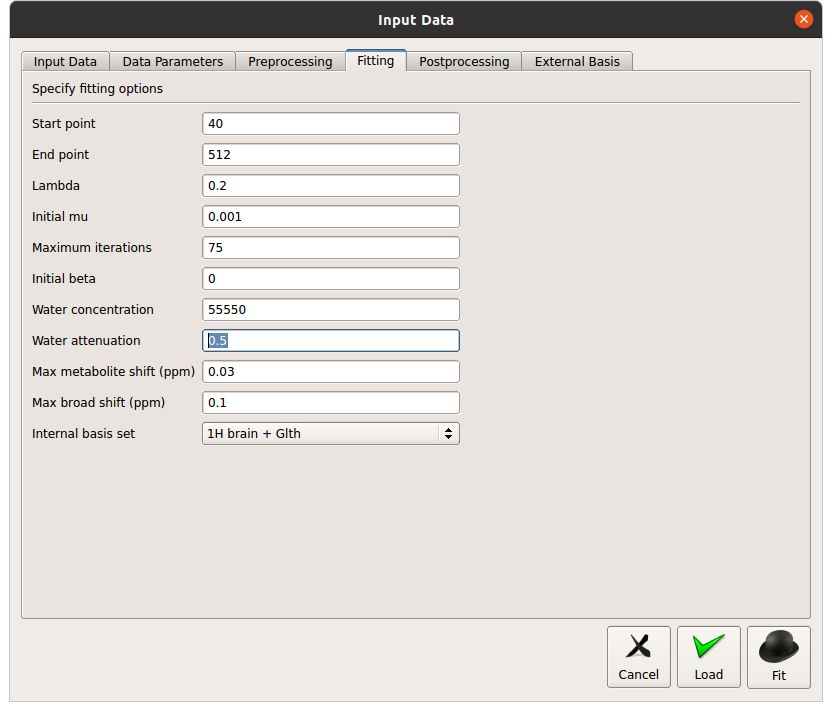
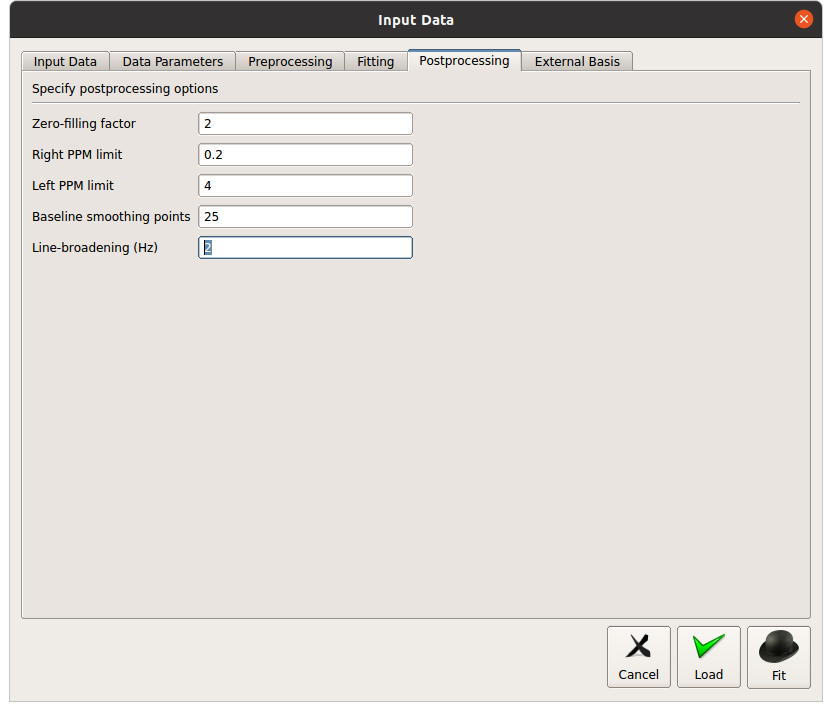
I didn’t understand what you mean by sparse?..I’m using version11 linux through GUI…and I have been using Hbrain+Glth…dod you mean I have to change it to hbrain?
On tarquin csv there is a number for water FWHM and residual did you see it?..
millions of thanks
Hi Neda,
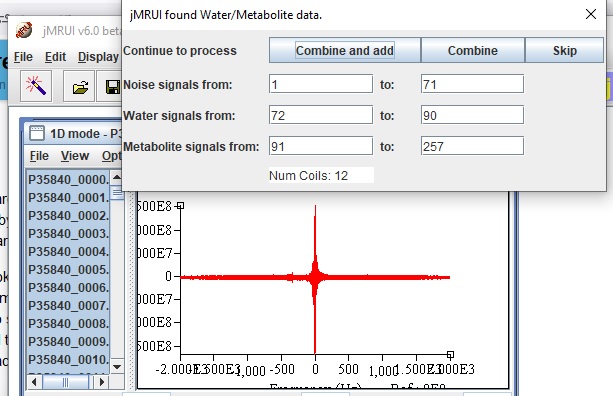
I suppose that this is the coil-combination routine in jMRUI (calling expert @amirshamaei in for confirmation). Did jMRUI populate the numbers in this window by itself or did you enter them? Which of the ‘continue to process’ options did you click?
Georg
Dear Georg,
Hi,
This is JMRUI beta 6 version and is done by itself. Jana had told me before that the automatic coil combination for MRSI GE is implemented in version 7 and in this version it’s still manual. I chose only combine option as I wanted to have water spectrum…
Neda
Hi Neda,
jMRUI
I’ve never tried jMRUI for MRSI data, so hopefully someone more experienced with that tool can step in – but: “Noise signals from: 1 to: 71” looks suspicious. I’m not sure whether jMRUI derives this from the header or attempts to infer it from the data, but I would not expect 27% of your acquired FIDs to be noise.
(water) fit and referencing:
Not all of the tools out there have been extensively tested with the newest software revisions and sequences; the most recent versions of Tarquin work well with single-voxel data from our MR750 at least up to DV26, but I’m less sure about MRSI and newer software releases.
The Tarquin fit you provided gave no indication of having found a meaningful water reference: all concentration estimates in your output are quoted in arbitrary units (au, not scaled to water amplitude), and the CSV file states: water amp 1, water FWHM (Hz) -1, water freq 0. These look like placeholder values, with the -0.0078 value you originally commented on being that -1 Hz, converted to ppm units.
If the water reference continues to be problematic, you may find it more practical to forgo water referencing and instead report concentrations relative to another metabolite (eg tCr) in this instance (see discussion here, for example)
Basis Set:
Regarding the basis set – the output you supplied shows only Cr, GPC, NAA, NAAG and PCh having been considered in the fit, and strong residuals (unsurprisingly) in other parts of the spectrum, most notably where Glu/Gln should be. There are many more metabolites in the 1H brain (+Glth) basis, but perhaps these were deselected in the GUI…
There’s a lot to process here – hopefully this sends you in the right direction, and isn’t just confusing…
Alex.
@alex @admin
Thank you very much for your time and kind attention…Regarding the internal basis set well I made another file just including Cho,GPC, NAA and GNAA Cr because my experiment is just a technical one about MRSI and I only need the main peaks and I preferred to have total ones but now I yet don’t know how can internal basis set affect my analysis while I’m choosing an external one…and about water information honestly I needed them as quality control parameters I preferred to have them… ![]() …Do you mean it;s not acquired actually??..and any recommendation you have for my analysis??..my experiment is quality control parameters comparison between two acquisition methods of MRSI related to grid…
…Do you mean it;s not acquired actually??..and any recommendation you have for my analysis??..my experiment is quality control parameters comparison between two acquisition methods of MRSI related to grid…
Thanks
Neda, omitting metabolites (like Glu) from the basis set will affect quantification of the ones that are included (like NAA). You should invest some time upfront to get the analysis right in the first place (even if it’s just ‘a technical experiment’), and understand precisely what and why is happening during your analysis. Can you share a screenshot of your Tarquin setup so we can check for other options?
Regarding the water scan, I agree with @alex, I tripped over the 71 noise scans as well. Do you have a copy of the acquisition protocol that you could share?
As @admin just mentioned: even if you’re only interested in reporting a few metabolites, it’s important that your basis set is reasonably “complete”, in that it covers all the metabolites you expect to see in the spectrum. A typical starting point might be:
Ala, Asp, GPC, PCh (Cho), Cr, PCr, GABA, Glc, Gln, Glu, GSH, Ins, Lac, NAA, NAAG, PE, Tau (ref)
Otherwise, you’ll find that signals from other (partially-overlapping) metabolites get lumped into the few components you have, and the baseline model may end up heavily contorted in an attempt to take up everything else (the latter is not such an issue with Tarquin, but will be in many other algorithms). You can later sum the outcomes to get the “total” estimates (Cr+PCr, PCh+GPC, NAA+NAAG).
Tarquin’s internal basis set is really convenient and can be a good starting point – you should be able to get better fits with an external basis, but only if it’s adapted to your exact sequence.
I have my doubts… but you’d have to check with someone more familiar with your sequence implementation, or try to figure it out by inspecting the individual FIDs (jMRUI is a great tool for that)
Opinions may vary, but I don’t think reporting the linewidth of water is critical in this context (although it may be preferable). If the data isn’t there (or the tools can’t find it), it’s not a disaster. Linewidth of (one or more of) the major metabolite peaks (eg, NAA or Cr) is likely to be more significant in determining how reliably the spectra can be decomposed…
Dear Georg,
Absolutely you are right…and honestly I didn’t intend to omit anything because of time!..I have attached the screenshots…Thanks alot Georg…
My acquisition protocol is TE=144/ TR=1000/voxel size:2.34 cc/ NEXT=1/ time:4/5 min/
Sorry for my late reply. jMrui finds the dataset’s max and min values, then calculates the mean, etc. Then, it performs a brute force search and determines water signals. It also employs a similar approach for noise signals.
@alex
Hi,
Sorry I had missed your reply and waiting for it! thank you very much for the explanations.
About the basis set how about considering only the long TE metabolites in an external basis set? when we know that for example we can’t have some of the metabolites in long TE like Asp or GLX if I’m right…or you still recommend the full basisset?.. what do you mean by exact sequence? its PRESS(probe) 3T GE and the scanner doesn’t give the metabolites:GABA, GSH,Tau and PE…so Do I still have to include them?..
Dear Amir,
Thank you for your reply…I uploaded a multivoxel pfile in jMRUI to find out the water spectrum but it seems suspicious…what’s your opinion?..
Hi @amirshamaei : thanks for following up on this. In principle, could the spatial encoding in the MRSI sequence be confusing this process?
No worries. It is hard to guess it since jMrui uses a brute force searching algorithm, and its performance might be dependent on your data. Is there any chance to have a sample of your data?
Yes, full basis set. You’ll still get plenty of signal from most metabolites at TE = 144 ms.
Yes, I will send you a sample thanks a lot Amir