Will you add support for import of MRS data in GE’s ScanArchive format to Osprey in the near future? Our now scaner does not save spectra in the older P file format.
Hi @joha09,
Thanks for reaching out.
This is definitely on our to-do list, as we are aware of the new format. However, we were so far unable to get our hands on a test dataset.
Would you be able to share the data and information about the acquisition with us so that we can work on the implementation?
Best,
Helge
Dear Helge,
Thank you for your prompt and enthusiastic reply. I am including phantom data sets obtained with two variants of the MEGA-PRESS sequence as it is implementet in GE’s AMRSP package, called GABAplus and GABA. The spectra don’t quite look like what you would expect from a human brain with measurable levels of GABA but I hope they can still be used to work out the structure of the new GE MRS format. I am also including the first three pages of a document that describes what pulse sequence parameters were used for the two sequence variants. The data are in the two zip files.
Best regards,
Jon
https://drive.google.com/file/d/1CEfwsAMgxq5oeQfgzeE8VyRwyfNqz4AC/view?usp=sharing
@noeskera I think the time has come where we might have to rely on your help. Also tagging @wclarke - this is the new GE file format I told you about. Maybe this is a good case where Ralph can help get conversion into spec2nii.
Still haven’t found the time to look into support for scan archives. I quickly converted the 2 scan archives into pfiles and analysed with GANNET. They look like editing spectra but just without any GABA. Strange. Need to think about a way to give you access to those pfiles.
But as long as we haven’t found a solution the site should work with pfiles. They are still saved automatically so I can’t understand the statement that they are not saved anymore. In addition to the standard /usr/g/mrraw location AMRSP copies pfiles into /usr/g/insite/tmp/exam#/series#.
Hope January I will find the time.
P45056_vox1_load.pdf (843.2 KB)
P45056_vox1_fit.pdf (313.4 KB)
P46080_vox1_load.pdf (852.9 KB)
P46080_vox1_fit.pdf (315.4 KB)
1 Like
Thanks Ralph! Fabulous (this is phantom data so no GABA).
@joha09 can you check in the second output folder Ralph mentioned, there should be p files in there you can use for now.
Dear Ralph,
Thank you for your input so far. I have located the p files on the scanner and put them in a zip file. The two files in folder 1276 are from the same phantom scan as before. They look like normal phantom scan files when I impor them into Osprey. The other folder (folder 1332 contains 4 p-files from scanning a volunteer with MEGA PRESS sequence, two locations in her brain, two scans for each location (GABA and GABA plus. When I import these files into Osprey two of them look like extremely noisy spectra files with no quantifiable resonances, while the other two files look like FID signals of pure water. It looks as though the scanner has used two different methods for generating the P files, one correct (the phantom) and one incorrect (the volunteer. Please take a look at the data and tell me if it looks like we have made som basic mistake when the volunteer was scanned. The zip file with all the p-files is in my Google drive at
https://drive.google.com/file/d/1NuyRAnBlXEH-_TSgqapDXiMvXMKc_7xG/view?usp=share_link
Best regards and thanks for your help on this.
Hi Jon,
Before we download data (please de-identify before sharing), can you post the Osprey outputs and the job file you used? (You’ll need different job files for the two sequences - we’ll need to simulate the MM-suppressed one for sure).
Thanks,
Georg
I will post a zip file named joha09.zip with the requested data, including the p-files with no personal information about the volunteer, job files generated by the Osprey GUI and all output generated by the analyses.When you unzip joha09.zip you will find that it includes 6 zip files. 7.zip and 8.zip are from the phantom scan that I posted earlier. Zip files 5, 6, 9 and 10 are from the volunteer scan. The intention was that files 5 and 9 would be from the GABA sequence and that files 6 and 10 would be from the GABA+ sequence. Here something went wrong: The p-files in 5.zip and 6.zip look like extremely noisy specta with no quantifiable resonances when they are imported into Osprey, while the p-files in 9.zip and 10.zip look like FID’s of a single peak (water) when imported into Osprey. I have no idea what is goint on here, since the same pulse sequences with the same parameters were used in both the successful phantom scan and the failed volunteer scan. I analyzed the data in Osprey one spectrum at a time.
https://drive.google.com/file/d/1_kq0ooEKgPTyeXkTxPzajG3TCPJ0f68p/view?usp=share_link
The error turns out to be reproducible. We have repeated the scan protocol with another volunteer (me this time). The result was exactly the same kind or weird p-files (two noisy “spectra” with no quantifiable resonances and two water FID’s).
Hi @joha09,
The raw p-file headers need special attention here; they will typically contain many of the subject details (including ID, date of birth etc) as originally entered on the console – some of the typical approaches for retrospectively removing these details from imaging data won’t apply to the raw p-files. There are a couple of readily-available tools out there for handling this offline, see for example: GitHub - schorschinho/MRSDeIdentificationTools: Tools to remove protected health information (PHI) from magnetic resonance spectroscopy data.
You may also be able to use the pfile_anon command on your scanner console (I’m not sure whether this is available on every system though)
…but back to your actual queries:
This all looks basically correct:
- Series 5 and 9 are the TE=80ms MM-suppressed implementation (20ms edit pulse at 1.9/1.5 ppm)
- Series 6 and 10 are the standard TE=68ms GABA+ implementation (15ms edit pulse at 1.9/7.46 ppm)
So the basic configuration looks reasonable, BUT… both Osprey and my own reader struggle with this, in much the same way. Latest Gannet does better (odd since at first glance it’s doing the same thing), but still seems to read some spurious points at the end of the FID which may indicate problems.
It looks like the raw data is arranged slightly differently from in other implementations (seemingly also with a bit more padding than the reader expects)… one thing I notice is a slightly unusual receiver count is reported (27 for some sequences, 44 for others); what coil are you using? This may be a factor, but I don’t think it’s the only issue…
I couldn’t quite get to the bottom of it today – perhaps Ralph @noeskera has some thoughts on the data ordering on this system, otherwise I’ll try to revisit this after Christmas ![]()
– Alex.
1 Like
Hi @joha09,
My guess is that you’re using the 48-channel head coil (AIR), with automatic element selection. I don’t have experience with MRS on these coils, but I suggest you could try the same sequences but manually selecting all 48 elements, and see if that works any better?
It looks like the raw data allows space for the full set of coil elements, but only fills the part selected. This seems to get confounded a bit by subsequent re-ordering under slightly different assumptions. I’ll try to follow up with a workaround in the reader function…
Alex.
I’ve updated Gannet’s GERead to handle this scenario; please see: Gannet GERead.m patch · GitHub (not tested very extensively, but it worked for Jon’s datasets and shouldn’t impact compatibility with any of the existing layouts).
[ UPDATE a few days later: this suggested workaround appears not to work correctly in all cases; needs some further development before use! ]
It should be pretty straightforward to update Osprey’s GELoad.m in much the same way – basically just need lines 267-324 and 382-384 from the updated file (after removing references to MRS_struct.p). I can do this sometime next week if you don’t beat me to it…
– Alex.
1 Like
Thank you for your work on this issue so far. I have just come back to work after a holiday break.
I am not sure how important this information is, but I have discovered that the issue with importing GE data into Osprey seems to be specifc for spectra generated by the Advanced MRS package. We did a scan of the MRS phantom with the standard PRESS sequence (PROBE) and another one with the GABA and GABAplus variants from AMRSP and tried to import both into Osprey. Import of PROBE spectra worked fine whhile the GABA/GABAplus spectra turned out to be impossible to import. This came as a surprise to me at least.
/Jon
https://drive.google.com/file/d/12aap6cQtebIcyDYmvzCWf7O3j3NNxQKq/view?usp=share_link
https://drive.google.com/file/d/13KRKs1Q7sNqSICHgnsdbObs4Ooz4kx0q/view?usp=share_link
I’ve pulled the changes into the Osprey GE reader function (thanks @alex !!!) and committed to the develop branch. It can load everything now, but I still don’t like how these data look.
(EDIT 1/5/23: I reverted that commit)
Alex, can you post the Gannet output for this? I’m mostly interested in the weird features that seem to be introduced by the water suppression:
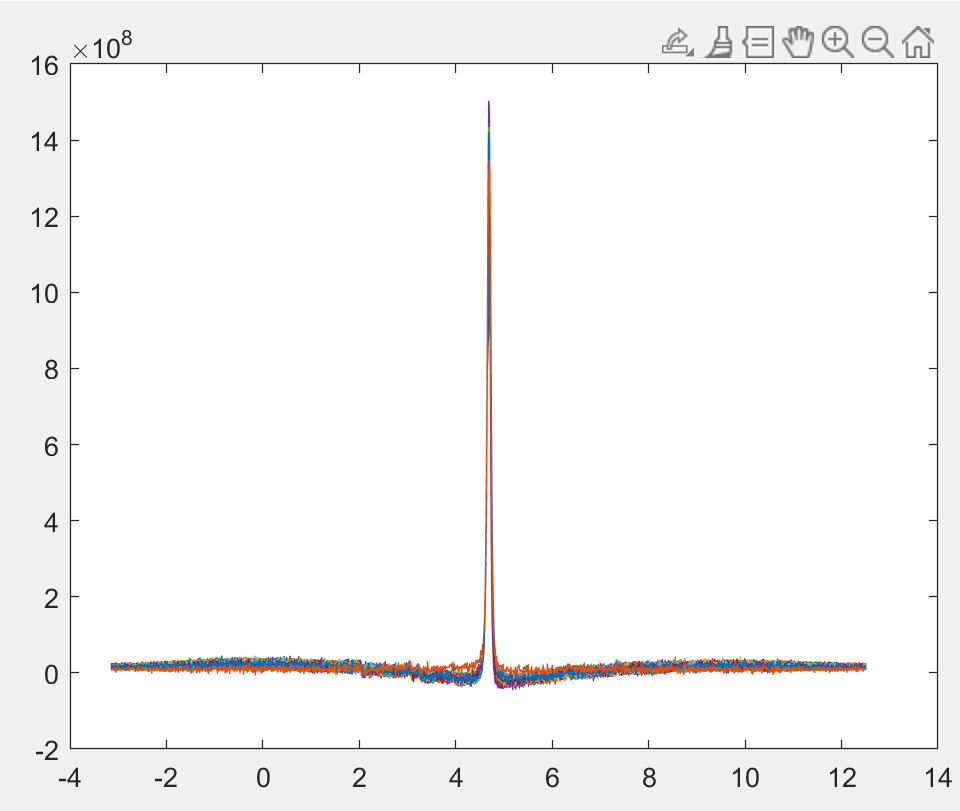
Osprey really struggled with the residual water removal because of this funky baseline bend. Jon, what are the water suppression settings here? @noeskera have you seen this before?
Dear All, sorry for looking into this so late (end of year is always super busy).
I checked series 5,6,9 and 10 and found all corrupted pfile.
As Alex pointed out you have most probably acquired the data with our automatic coil selection mode (Air Touch). This de-selects low SNR coil elements automatically. Unfortunately, this results in 99% of the cases in corrupted data files (sometimes very obvious).
The only workaround in the moment is to use manual coil mode which works only in research mode (you have to be using the WIP) and using the wheel in the upper right hand corner of the coil selection window.
You should then always get the full number of coil elements (otherwise don’t expect to get un-corrupted raw data). To be on the safe side you can also store the scan archives as I can create correct pfiles from those (I had a couple of cases where the workaround didn’t work. We never found the reason for that).
This usually doesn’t happen with phantom as we tend to place the vocal in center and none of the coil elements get de-selected by Air Touch.
So you have to re-acquire your data with manual coil mode and that should fix both your in-vivo data quality and all the reading issues.
The problem and workaround was only reported to our Apps people and on our collaboration portal (so not all are aware of this issue).
It will be fixed with the next software release (MR29.1_R06 and MR30.0).
You can also get directly in touch with me (ralph.noeske@med.ge.com).
Sorry again for the inconvenience and trouble this caused to all of you.
Cheers and have a great 2023.
3 Likes
Thanks for the clarification @noeskera – I recall you had previously mentioned some complications with Air Touch, but was uncertain whether this was by design or otherwise.
I looked a little more closely at my workaround, and can see that in some cases there’s still an off-by-one error in where the reader is reading, for some of the FIDs – this could very well result in a slowly undulating baseline component after fft, which might be what you see here @admin (it also gives rise to poor SNR in some of the reconstructed spectra).
If these are considered “corrupt” and have been fixed in subsequent versions, then perhaps it’s not worth further pursuing a fix in the reader at this stage – but keep it in mind as a possibility if someone needs it to salvage an entire study? (@joha09, as I understand it you’re just piloting at this stage – so a couple of lost datasets would not be a disaster)